|
|
|
Le métissage entre les premiers humains
Les secrets cachés dans notre ADN (I) Notre patrimoine génétique est le résultat de millions d'années d'évolution. Bien que nous le sachions, tout le monde fut surpris d'apprendre en 2006 que nous possédons encore quelques gènes actifs que nous avons hérité des hommes de Néandertal (430000-26000 BP). Certaines populations ont même hérité de gènes de Dénisoviens (48000-29200 BP) et peut-être d'autres variétés humaines plus archaïques et encore inconnues. Voyons ces découvertes en détails. Il y a du Néandertal en nous Jusqu'au début des années 2000, les paléontologues ne disposaient que de neuf tests ADN de Néandertaliens. Ces informations génétiques restent donc rares et précieuses. L'ADN mitochondrial d'un dixième néandertalien, celui extrait d'une molaire de l'enfant de Néandertal fut analysé en 2006 par le laboratoire de biologie moléculaire de l'ENS de Lyon. Les résultats confirment que le fossile est bien âgé de 100000 ans et donc la présence de Néandertal avant l'arrivée de l'Homo sapiens (il y a 315000 ans) et leur cohabitation. L'analyse ADN a également appuyé la thèse selon laquelle le patrimoine génétique de l'homme de Néandertal s'est appauvri au fil du temps. Enfin, cette analyse a prouvé sans équivoque que l'homme de Néandertal appartient à une espèce différente de l'Homo sapiens. En d'autre terme, l'homme de Néandertal n'est pas notre ancêtre direct. Toutefois, nous savons qu'en biologie rien n'est binaire, noir ou blanc, et que dame Nature est très subtile, bien plus imaginative que nos ingénieurs les plus habiles. Des chercheurs ont donc approfondi la question et cherché à savoir si des croisements avaient pu se produire entre l'homme de Néandertal et l'Homo sapiens à l'époque où les deux espèces vivaient sur les mêmes terres. Avant cette analyse rien ne permettait de l'affirmer. Mais les chercheurs qui ont étudié le matériel génétique de l'enfant de Néandertal étaient unanimes pour dire que ce n'était pas exclu. Leur point de vue sera confirmé par l'analyse d'autres fossiles quelques années plus tard. En 2010, une équipe internationale de généticiens et de paléontologues dirigée par le généticien suédois Svante Pääbo de l'Institut Max-Planck (EVA) mit en évidence que le génome des populations non-Africains contient en moyenne 2% (entre 1 et 4%) d'ADN néandertalien. Deux études publiées dans les revues "Science" en 2014 et "Nature" en 2021 confirment que collectivement, les humains modernes possèdent environ 20% d'ADN néandertalien. La localisation de ces gènes est différente chez chaque individu et ils ont divers impacts : dans la couleur des cheveux, la couleur de la peau et les maladies. Cette surprenante conclusion fait suite à la découverte d'os appartenant à trois femmes de Néandertal vieilles d'environ 38000 ans dans la grotte de Vindija, en Croatie vers 1980. Plus récemment, les chercheurs ont découvert que le génome des populations de souche mélanésienne contenait plus de 5% de génome archaïque, soit plus du double des Occidentaux. On y reviendra (cf. l'Homo denisova).
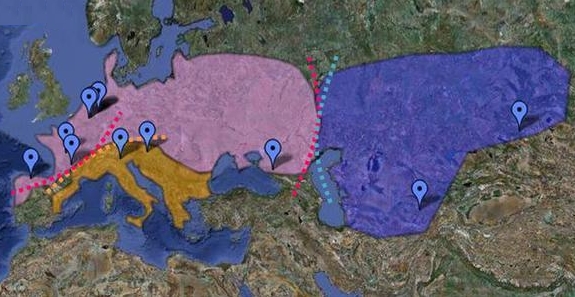
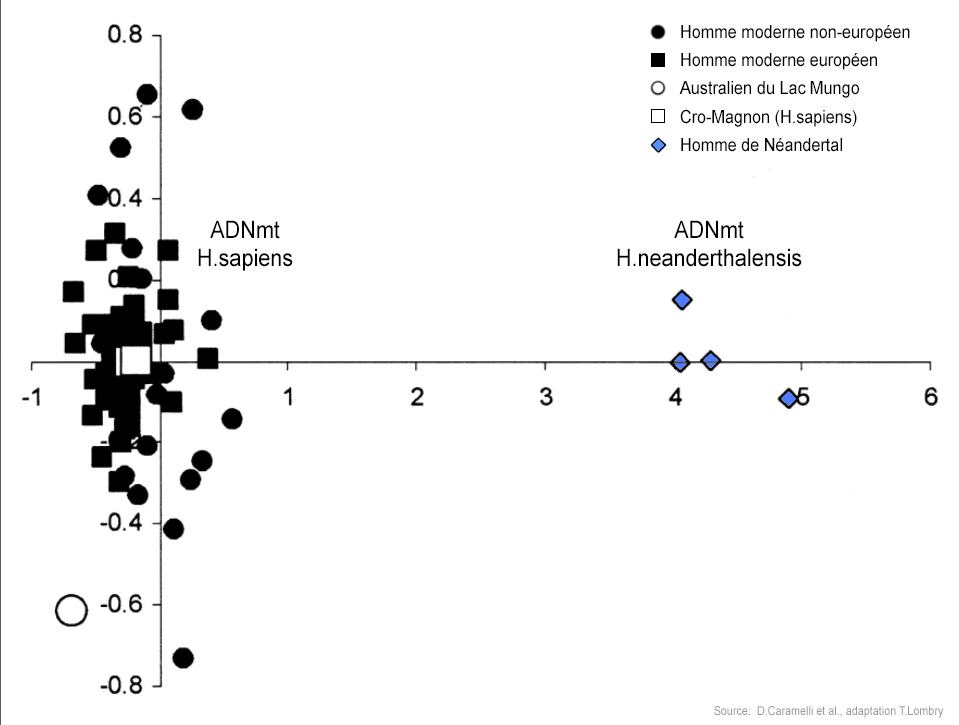
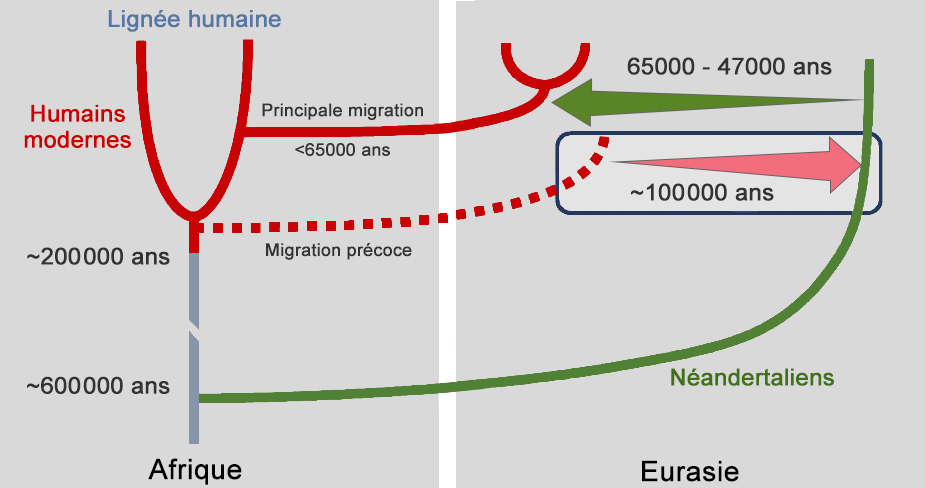
En 2006, Svante Pääbo avait annoncé qu'il voulait reconstruire l'ADN de l'homme de Néandertal, ce qui donna naissance au "Neanderthal Genome Project". Au cours de leurs analyses, les chercheurs ont comparé l'ADN de cinq individus Européens (Français), Chinois, Papous, d'Afrique de l'Ouest (Yoruba) et d'Afrique du Sud (San) et constaté qu'il est identique à 99.7% à celui de l'homme de Néandertal. Mais sur les 5 ADN étudiés, celui des Africains est légèrement plus éloigné des Néandertaliens que les autres. Autrement dit, les Européens, les Chinois et les Papous se sont génétiquement "rapprochés" des Néandertaliens après avoir quitté l'Afrique. Selon Svante Pääbo, cette proximité génomique n'a qu'une seule explication : c'est la preuve qu'il y eut des croisements entre Néandertal et Homo sapiens. Si les Néandertaliens sont présents dans toute l'Eurasie, appartiennent-ils tous à la même espèce ? D'expérience on peut en douter vu leur vaste distribution géographique. En 2009, des études de l'ADN mitochondrial conduites par Virginie Fabre du Laboratoire d'Anthropologie Bio-culturelle du CNRS de l'Université de Méditerranée à Marseille et ses collègues ont permis de confirmer l'existence d'au moins 3 sous-groupes de Néandertaliens et d'un possible 4e en Asie. Ceux-ci sont distribués entre l'Europe occidentale, le bassin Méditerranéen et le Proche-Orient comme le montre la carte ci-dessus. Les contacts et le métissage entre les deux populations d'homininés ont donc dû se produire hors d'Afrique, à l'époque où les différentes populations se sont séparées et colonisèrent l'Europe et l'Asie. Ainsi, grâce à l'analyse de l'ADN des humains, en 2016 une équipe de chercheurs dirigée par Martin Kuhlwilm de l'Institut Max Planck découvrit que ce métissage se serait produit une première fois des Homo sapiens vers les Néandertaliens il y a 100000 ans soit plus de 40000 ans plus tôt que prévu, puis de façon importante des Néandertaliens vers les hommes modernes entre 65000 et 47000 ans. Il y eut également des métissages au Proche-Orient il y a 80000 à 60000 ans. On ignore toutefois dans quelles proportions et à quelle fréquence les deux populations se sont métissées.
Si on estime que de 1 à 4% de l'ADN des populations non africaines actuelles provient des hommes de Néandertal, cela signifie que ces éléments génomiques sont exactement identiques à ceux de Néandertal. Le reste ? Les 96% restants forment les différences comme il en existe entre les membres d'une même famille possédant des gènes en commun. Ainsi, votre cousin partage environ 1/8e soit 12.5% de votre génome. Cela ne veut pas dire que 87.5% de vos gènes sont différents de celui de votre cousin. Tous les humains ont le même ADN à 0.1% près. Quelles particularités avons-nous hérité de Néandertal ? Actuellement on ignore quels sont ces 1 à 4% de gènes que nous a légué Néandertal. En fait, ces morceaux de gènes sont distribués au hasard sur l'ensemble de l'ADN et on commence seulement à les identifier et à comprendre leur rôle. La comparaison gène à gène a déjà permis d'obtenir certains résultats. 15 loci ou régions du génome humain comprennent entre 1 et 12 gènes "sélectionnés positivement", selon Svante Pääbo. Ainsi, le gène THADA, impliqué dans le métabolisme des cellules aurait donné à l'homme moderne un supplément d'énergie (cf. R.E. Green et al., 2010). Le gène HLA a renforcé nos défenses immunitaires (cf. R.E. Green et al., 2010) alors que le gène TLR nous rend plus sensible aux allergies, notamment à l'asthme et au rhume des foins (cf. J.Kelso et al. 2016). Le gène RUNX2 qui joue un rôle dans la formation et la suture des os aurait donné à l'homme moderne la forme de son crâne ou les dimensions du thorax (cf. S.Pääbo et al., 2013). D'autres gènes ont modifié notre pilosité ou favorisé la synthèse de la vitamine D malgré le faible ensoleillement. En revanche, d'autres gènes hérités des Néandertaliens vivant en Croatie sont associés à un taux élevé de cholestérol, à la schizophrénie et à l'arthrite rhumatoïde (cf. S.Pääbo et al., 2017). Entre 1.5 à 7% de notre génome est unique D'un autre côté, notre génome possède des régions continues qui sont totalement dépourvues d'ADN néandertalien. Entre seulement 1.5 et 7% de notre ADN est unique à l'homme moderne (cf. N.K. Shaefer et al., 2021). En étudiant ces gènes spécifiques à l'Homo sapiens, les généticiens espèrent en apprendre un peu plus sur nous-mêmes. Nous avons par exemple des preuves que de multiples explosions de changements adaptatifs spécifiques aux humains modernes sont apparues au cours des 600000 dernières années impliquant des gènes liés au développement et à la fonction du cerveau (cf. N.K. Shaefer et al., 2021). Ces régions comprennent notamment des gènes tels que FOXP2, qui est impliqué dans la coordination motrice et pourrait jouer un rôle important dans le langage et la parole humaines. On a également trouvé des changements dans le chromosome X où des gènes semblent réduire la fertilité des hommes. Parmi 1500 gènes impliqués dans le système immunitaire, il apparaît que la plupart des adaptations se sont produites entre 13000 et 6000 ans, quand les derniers chasseurs-cueilleurs sont passés à l'agriculture (cf. L.Qintana-Murci et al., 2016). Vers une révision du concept d'espèce Mais s'il y eut des croisements entre Néandertal et Homo sapiens, cela signifie que les deux soi-disant "espèces" étaient compatibles, viables et fécondes. Si cette hybridation est fertile - nous sommes-là pour en témoigner - cela signifie donc que contrairement à ce qu'on pensait (notamment en étudiant uniquement l'ADN mitochondrial) les deux embranchements forment une seule et même espèce. A moins que notre définition d'une espèce soit incomplète et qu'elle englobe des concepts que nous ignorons encore.
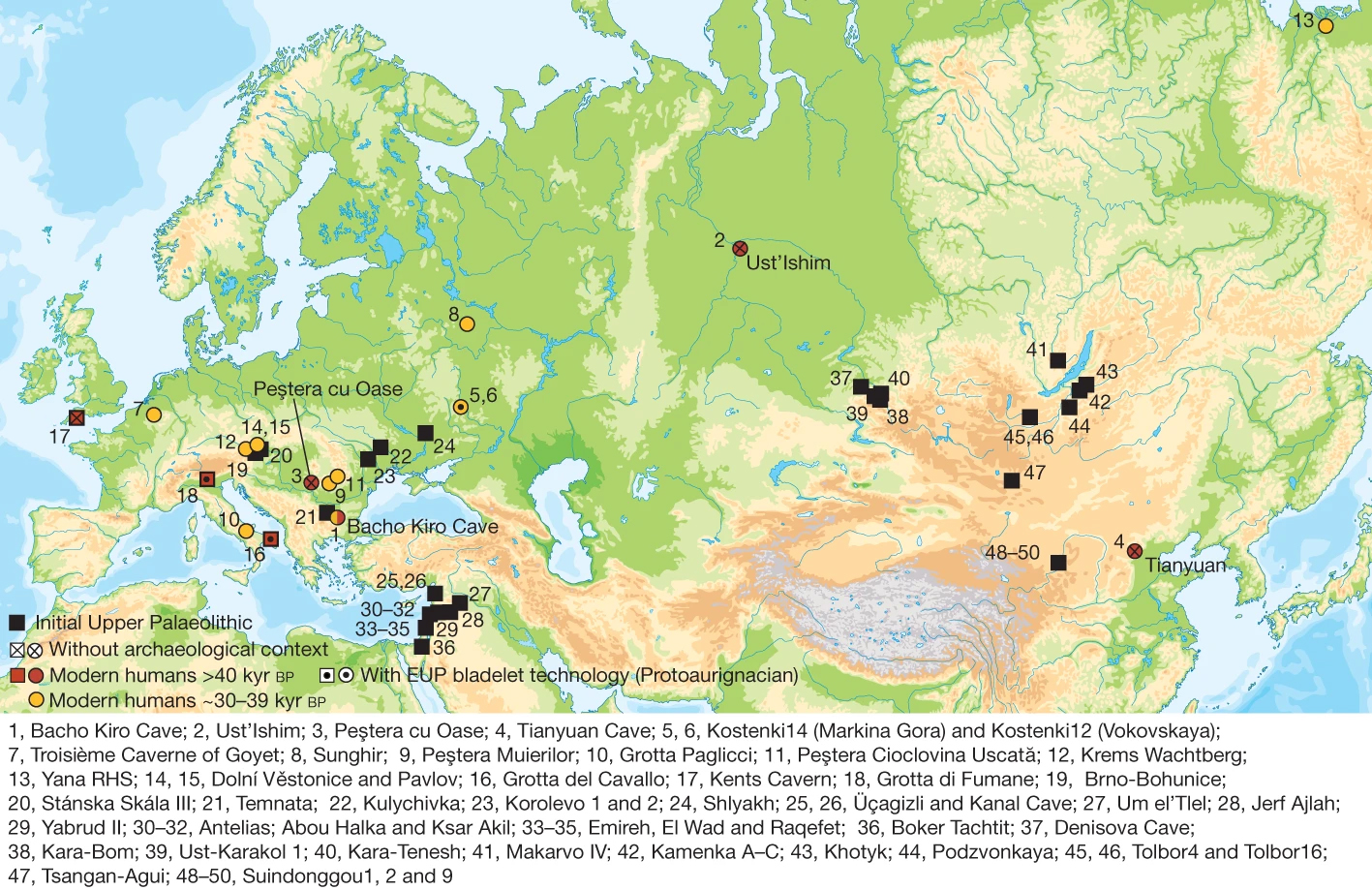
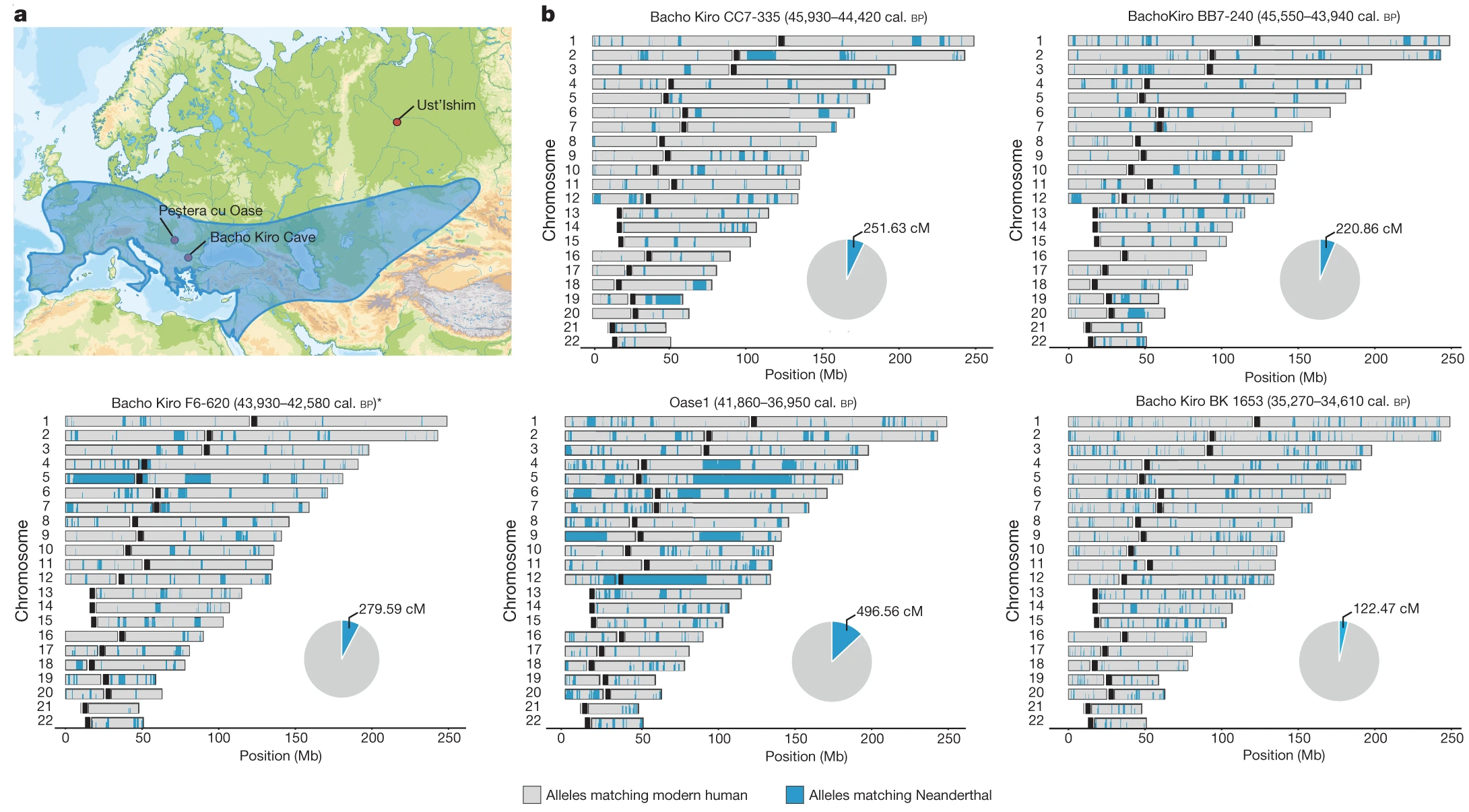
Nous savions déjà que les deux groupes d'homininés avaient cohabités : Néandertal et Homo sapiens ont vécu 50000 ans côte-à-côte au Proche-Orient et plus tard dans le sud de l'Europe. On a également retrouvé des outils et des objets tellement semblables qu'il est impossible de les attribuer à l'une ou l'autre population. La conclusion est donc évidente : soit les deux populations se sont rencontrées soit il y eu convergence. Pour appuyer la première hypothèse, rappelons qu'en 1998 un squelette métissé d'enfant fut découvert au Portugal (Lagar Velho); à la fois Néandertalien (par la robustesse de ses os et de ses mandibules) et Homo sapiens (par l'anatomie de son crâne, ses dents, radius et pubis), on ne peut pas le classer dans une espèce précise, à moins qu'il s'agisse d'un juvénile aux caractères encore mal définis. Des hybridations en Europe il y a au moins 219000 ans Dans tous les cas, des croisements entre Néandertal et Homo sapiens ont donné une descendance fertile, des petits H.neanderthalensis sapiens il y a plus de 80000 ans, et en nombre suffisant pour laisser des traces dans notre ADN. En fait, cette hybridation commença bien plus tôt. En effet, en 2017 Cosimo Posth et ses collègues ont publié dans la revue "Nature" des preuves qu'il y eut des brassages génétiques de Néandertaliens en Europe (Allemagne) il y a 460000 à 219000 ans. En 2016, Martin Kuhlwilm et ses collègues ont également annoncé dans la revue "Nature" avoir trouvé des traces génétiques indiquant que des groupes pionniers d'Homo sapiens africains se sont hybridés avec des Néandertaliens dans la région de l'Altaï en Sibérie il y a environ 100000 ans. Depuis les années 1950, on a également découvert des métissages entre les Néandertaliens et les premiers Homo sapiens vivant en Eurasie. Il y a des exemples dans la grotte de Bacho Kiro en Bulgarie (des os datés entre 45930-42580 ans BP), dans la région de Zlaty Kun (un crâne de 45000 ans BP) en Tchéquie, en Roumanie (le spécimen Oase 1 daté de 42000-37000 ans BP) et en Sibérie (le spécimen de Ust-Ishim daté de ~45000 ans). Les analyses génétiques des restes humains (dents et os) prouvent que des Homo sapiens avaient des ancêtres néandertaliens remontant à cinq ou sept générations, suggérant que les métissages entre ces premiers humains d'Europe et les Néandertaliens étaient courants (cf. M.Hajdinjak et al., 2021).
Etant donné ces brassages interspécifiques (entre deux espèces différentes), il faut donc revenir sur la définition d'espèce telle que Buffon la proposait au XVIIIe siècle : "une même espèce est celle qui, au moyen de la copulation, se perpétue et conserve la similitude de cette espèce." Cette définition sous-entend que tout accouplement stérile interdit de classer les parents dans la même espèce (exemple du cheval et de l'ânesse dont le bardot est stérile). Inversement, que l'union de deux espèces ne peut pas engendrer de descendance fertile ni maintenir leur spécificité. En effet, depuis le milieu des années 1990 les biologistes ont relevé de nombreuses exceptions qui viennent mettre en pièce le concept d'espèce.. L'exemple le plus simple d'espèce hybride donnant une population féconde est celui du loup et du coyote du nord des Etats-Unis qui donne naissance à des individus fertiles qui restent deux espèces bien distinctes. La fondation coréenne SOOAM a également pu obtenir par insémination artificielle des coyotes fertiles nés d'une chienne porteuse. Il existe également quelques rares cas naturels de zébranes issus du croisement d'un zèbre et d'un âne. En théorie la progéniture est stérile mais quelques zoos en possèdent qui sont fertiles. Charles Darwin cite même le cas d'un zébrâne issu d'un triple hybride avec une jument. Il y eut également aux Barbades une poignée de zèbres-bardots (cheval-anesse) mais le jeune n'a pas survécu jusqu'à l'âge adulte. On en déduit que s'il est possible d'obtenir une descendance féconde entre espèces apparentées, il est impossible que des espèces hybrides (zébrâne et autre zébrule) donnent une progéniture viable. Toutefois certains chercheurs tentent prouver le contraire.
S'il n'était pas évident au début du siècle dernier de classer les espèces successives à l'origine des Homo sapiens modernes, aujourd'hui, alors que paradoxalement nous sommes épaulés par la génétique et l'informatique, la frontière entre les espèces est encore plus difficile à définir. Malgré les grandes avancées faites par les paléontologues, les généticiens et les biologistes, cette nouvelle découverte va forcer les scientifiques à revoir leur système de classification depuis le préhumain au faciès simiesque et écervelé jusqu'à l'homme moderne. Si cette découverte risque de relancer le débat dans l'esprit de certains opposants à la théorie de l'Évolution, elle tombe à point nommer pour agrandir les membres de notre petite famille. Un chromosome Y d'Homo sapiens chez les Néandertaliens Il est bien établi que toutes les personnes d'ascendance non africaine portent une petite quantité d'ADN de Néandertal à la suite du croisement entre les Néandertaliens et les Sapiens il y a environ 70000 à 50000 ans, peu de temps après que les humains modernes aient migré hors d'Afrique et aient commencé à se propager autour du monde. Cependant, la question de savoir si les Néandertaliens peuvent également porter un ADN humain moderne a fait l'objet d'un débat. Comme expliqué précédemment, on a longtemps cru que les Néandertaliens n'avaient jamais pu donner une descendance fertile à des Homo sapiens. Une nouvelle découverte vient contredire cette hypothèse non fondée. Dans une article publié dans la revue "Science" en 2020, une équipe internationale de généticiens dirigée par Martin Petr et Janet Kelso de l'Institut Max Planck d'anthropologie évolutive (EVA-MPG) de Leipzig, en Allemagne, séquença les génomes de trois Néandertaliens et de deux Dénisoviens en ciblant le séquençage sur le chromosome Y.
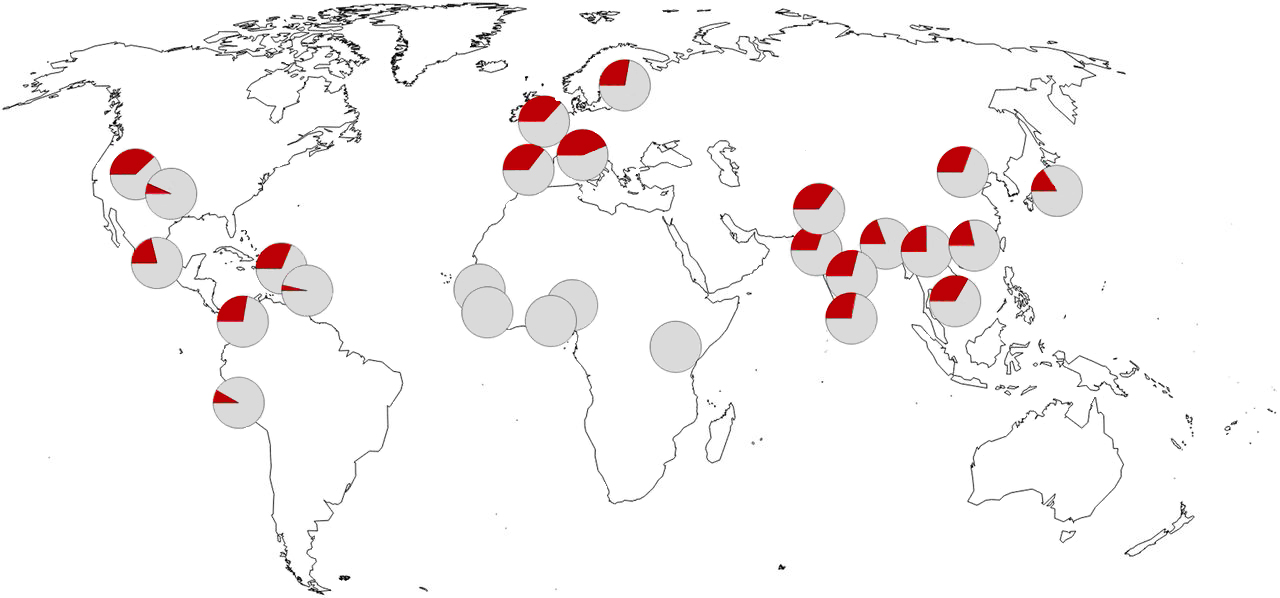
Pour rappel, le chromosome Y se transmet génétiquement uniquement de père en fils. Seuls les hommes disposent d'un chromosome Y et d'un chromosome X. Les femmes disposent de deux chromosomes X et héritent par leur mère de l'ADN mitochondrial. Ce chromosome Y semble être une relique ne jouant plus aucun rôle puisque les femmes s'en passent bien. On reviendra sur l'avenir incertain du chromosome Y à propos de l'avenir de l'Homme. Les chercheurs ont comparé les chromosomes Y des Homo sapiens à ceux des Néandertaliens et des Dénisoviens. Ils ont découvert qu'il y a plus de parenté entre que les chromosomes Y des humains et des Néandertaliens qu'avec celui des Dénisoviens. L'étude apporte les preuves que les Néandertaliens ont peut-être profité du métissage avec les Homo sapiens car le flux génétique entraîna le remplacement complet du chromosome Y d'origine Néandertalienne par son homologue Homo sapiens. Les chercheurs ont également calculé que l'ancêtre commun le plus récent du chromosome Y de Néandertal et de Sapiens remonte à environ 370000 ans, bien plus récemment qu'on ne le pensait jusqu'ici. Selon les chercheurs, ces séquences chromosomiques Y fournissent maintenant de nouvelles preuves que les Néandertaliens et les premiers humains modernes se sont rencontrés et ont échangé des gènes avant la migration majeure hors d'Afrique - potentiellement il y a 370000 ans et certainement plus de 100000 ans. Cela implique qu'une population étroitement liée aux premiers humains modernes devait déjà avoir été en Eurasie à cette époque. Étonnamment, ce croisement entraîna le remplacement des chromosomes Y d'origine des Néandertaliens par ceux des premiers humains modernes, un schéma similaire à ce qui fut observé pour l'ADN mitochondrial néandertalien dans une étude antérieure. Pour vérifier si ce remplacement n'était pas dû au hasard, les chercheurs ont réalisé des simulations informatiques et découvert que la petite taille des populations de Néandertal pourrait avoir conduit à une accumulation de mutations délétères dans leurs chromosomes Y qui aurait réduit leur aptitude évolutive. Ce phénomène est assez similaire aux situations dans lesquelles des populations extrêmement petites et la consanguinité peuvent parfois augmenter l'incidence de certaines maladies. Selon Martin Petr, "Nous pensons qu'étant donné le rôle important du chromosome Y dans la reproduction et la fertilité, la moindre aptitude évolutive des chromosomes Y de Néandertal pourrait avoir provoqué une sélection naturelle pour favoriser les chromosomes Y des premiers humains modernes, conduisant finalement à leur remplacement." Janet Kelso, autrice principale de cet article estime que cette hypothèse de remplacement pourrait être testée dans un proche avenir : "Si nous pouvons récupérer les séquences du chromosome Y de Néandertaliens qui vivaient avant cet évènement d'introgression précoce supposé, comme chez les 430000 an .des Néandertaliens de Sima de los Huesos en Espagne, nous prédisons qu'ils auraient toujours le chromosome Y Néandertalien original et seront donc plus similaires aux Dénisoviens qu'aux humains modernes." Des gènes à risque et protecteurs néandertaliens Des gènes à risque de Covid-19 L'héritage du patrimoine génétique de nos Ancêtres cause parfois plus de mal que de bien. Des études ont montré que certaines mutations génétiques étaient fréquemment présentes chez les patients Covid souffrant d'une forme sévère de la maladie. A ce jour, on a identifié 22 variants génétiques ou allèles touchant 8 gènes différents ayant un effet délétère sur la réponse du système immunitaire contre le Covid-19. Près de 15% des formes sévères de Covid s'expliquent par ces prédispositions génétiques (cf. Q.Zhang et al., 2020; Covid-19 HGI, 2020). Une mutation sur des loci à risque de Covid-19 du chromosome 3 fut observée chez des patients gravement malades traités en Italie et en Espagne (cf. D.Ellinghaus et al., 2020). Dans une étude publiée dans la revue "Nature" le 30 septembre 2020, le médecin chercheur Hugo Zeberg et le paléogénéticien Svante Pääbo de l'EVA-MPG ont montré que le risque est conféré par un segment génomique de ~50 kbp héritées des Néandertaliens (cf. L.Jin et al., 2013). Selon les auteurs, de nos jours cet haplotype (un groupe de gènes variables ou allèles de différents loci situés consécutivement sur un même chromosome) dont rs11385942 sur le locus 3p21.31 est presque complètement absent chez les populations d'Afrique. En revanche, il est porté par ~50% des Asiatiques du sud, ~ 16% des Européens, ~4% des Américains et par les habitants d'Océanie. Il est également suprenant de constater que ces loci à risque sont quasiment absents en Asie de l'Est. Ces loci étaient déjà présents chez les Néandertaliens qui vécurent dans l'actuelle Croatie il y a 50000 ans (cf. K.Prüfer et al., 2017). Cet haplotype est similaire à d'autres variants génétiques présentes chez les Néandertaliens et les Dénisoviens. Concrètement, la mutation déplace la cytosine (C) en position rs10490770 et la guanine (G) en rs35044562. Celles-ci sont en "déséquilibre de liaison complet", ce qui signifie que si vous avez C au premier endroit, vous avez presque certainement G au second. Chacun peut connaître son génome et rechercher C en rs10490770 (cf. le test génétique de 23andMe ou leur outil de recherche Browse Raw Data par exemple ou les fichiers de données brutes de Family Finder et Ancestry).
La fréquence la plus élevée apparaît au Bangladesh, où plus de la moitié la population (63%) porte au moins une copie des loci à risque Néandertaliens. Les auteurs confirment que "l'haplotype Néandertalien peut donc être un contributeur substantiel au risque de Covid-19 chez certaines populations en plus d'autres facteurs de risque, notamment l'âge avancé. A priori en accord avec cela, chez les personnes d'origine bangladaise du Royaume-Uni, le risque de décéder du Covid-19 est environ deux fois plus élevé que dans la population générale (probab. de 95%)." Cette grande différence de fréquences des allèles entre l'Asie du Sud et l'Asie de l'Est est inhabituelle (probab. 0.6%) et peut être le signe d'une sélection remontant loin dans le passé. En effet, des travaux antérieurs suggèrent que l'haplotype de Néandertal fut sélectionné positivement (surreprésenté) au Bangladesh (cf. S.R. Browning et al., 2018). Une raison serait qu'il offrirait une protection contre d'autres agents pathogènes. Il est également possible que l'haplotype fut moins fréquent dans l'est de l'Asie en raison d'une sélection négative (sous-représenté), peut-être liée à des virus de type Corona ou d'autres agents pathogènes. Dans tous les cas, les loci à risque de Covid-19 sur le chromosome 3 sont similaires à d'autres mutations génétiques qui ont atteint des fréquences élevées dans certaines populations, mais aujourd'hui sous-représentées en raison de la pandémie de Covid-19. Selon les auteurs, "On ne sait pas actuellement quelle caractéristique dans le variant des loci Néandertaliens confère un risque de Covid-19 sévère et si ces effets sont spécifiques au Covid-19, à d'autres coronavirus ou à d'autres agents pathogènes [...] Cependant, en ce qui concerne la pandémie actuelle, il est clair que le flux génétique des Néandertaliens a des conséquences tragiques." Des gènes protecteurs contre la Covid-19 sévère Si certaines populations ont hérité des Néandertaliens un facteur de risque génétique de développer une Covid-19 sévère, des études d'association génétique plus vastes permettent de découvrir d'autres facteurs de risque génétiques. Dans une étude publiée dans les "PNAS" en 2021, Hugo Zeberg de l'Insitut Karolinska de Stockholm et Svante Pääbo de l'EVA-MPG ont montré que d'autres gènes hérités des Néandertaliens ont cette fois un effet protecteur.
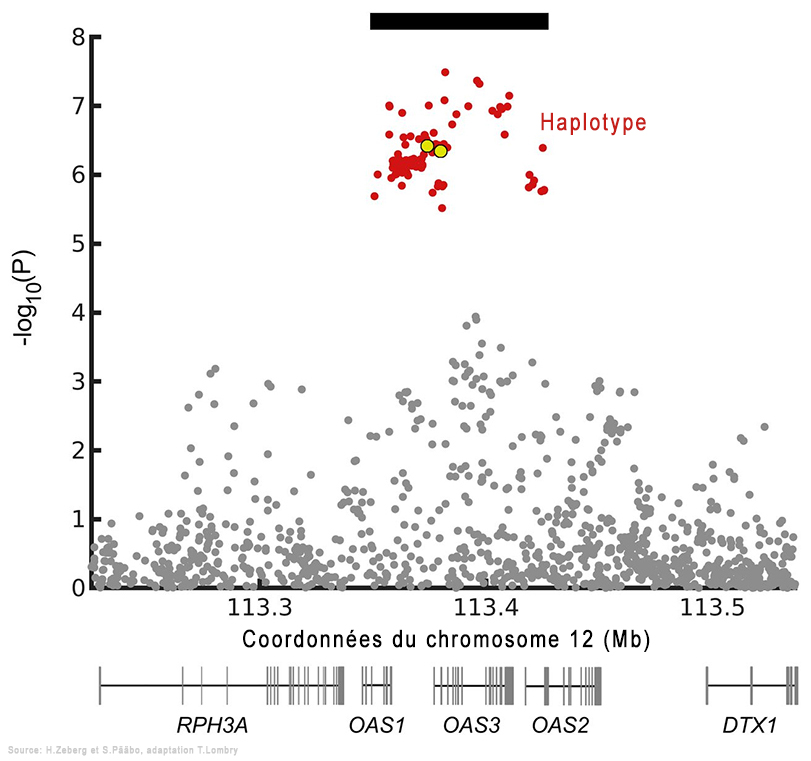
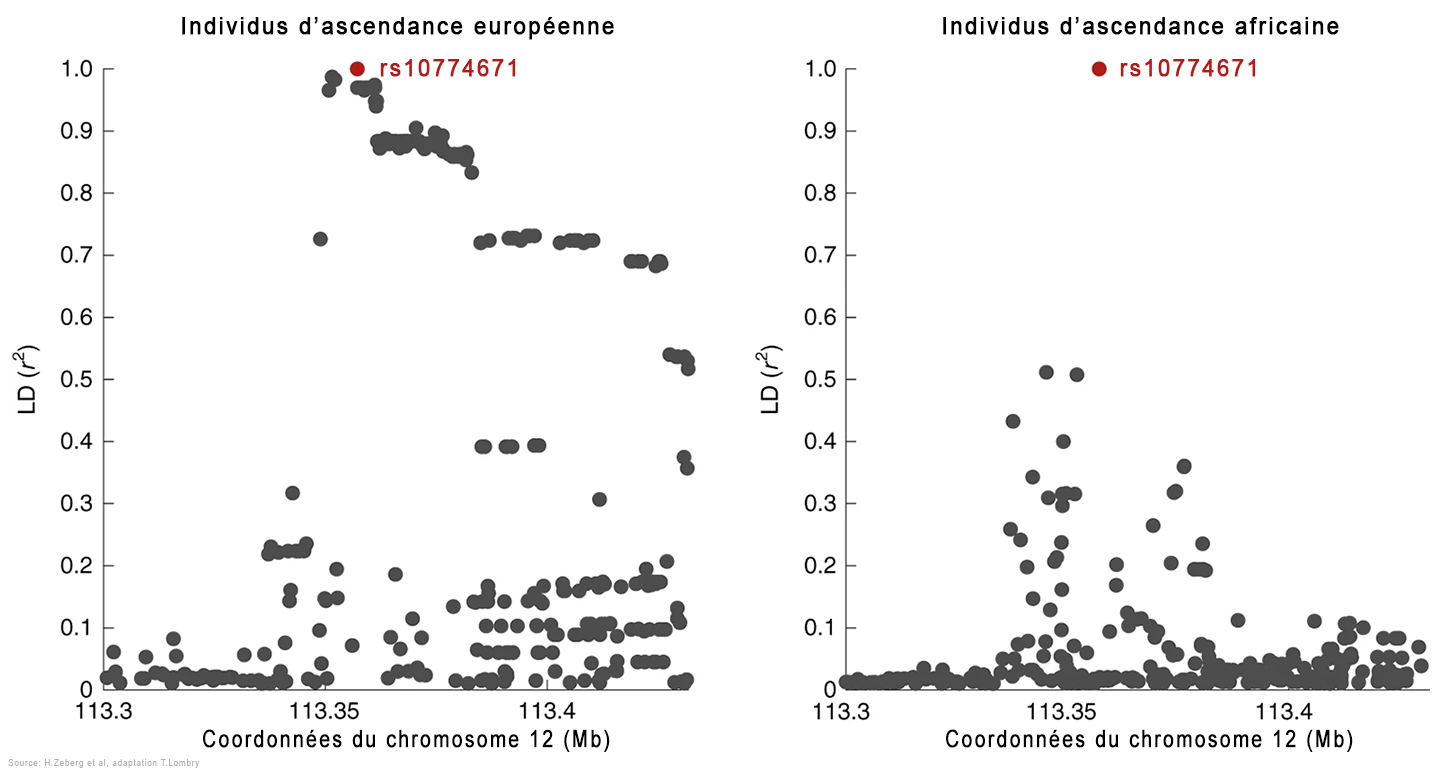
En utilisant les données du consortium Genetics of Mortality in Critical Care (GenOMICC), les chercheurs ont découvert qu'un haplotype (un groupe de gènes variables ou allèles de différents loci situés consécutivement sur un même chromosome) situé dans une région du chromosome 12, le locus OAS, est hérité des Néandertaliens. Lorsqu'il est contaminé par le Covid-19, le patient est hospitalisé aux soins intensifs. Cette région code pour des protéines qui activent des enzymes importantes lors d'infections par des virus à ARN. Mais contrairement à l'haplotype néandertalien décrit ci-dessus qui augmente le risque de Covid-19 sévère, cet haplotype néandertalien protège contre une forme grave de la maladie en réduisant de ~22% le risque de développer une Covid-19 sévère. Il diffère également de l'haplotype à risque en ce qu'il a un effet plus modéré et se produit plus fréquemment dans toutes les régions du monde en dehors de l'Afrique. Parmi les génomes humains anciens de l'Eurasie occidentale, la fréquence de l'haplotype protecteur néandertalien peut avoir augmenté voici 20000 à 10000 ans et à nouveau au cours des 1000 dernières années. L'équipe d'Hugo Zeberg a ensuite cartographié de manière précise le locus OAS1/2/3 chez 2787 patients Covid d'ascendance africaine et chez 130997 personnes d'un groupe témoin ayant fait l'objet de six études précédentes. Leurs résultats furent publiés dans la revue "Nature Genetics" en 2022.
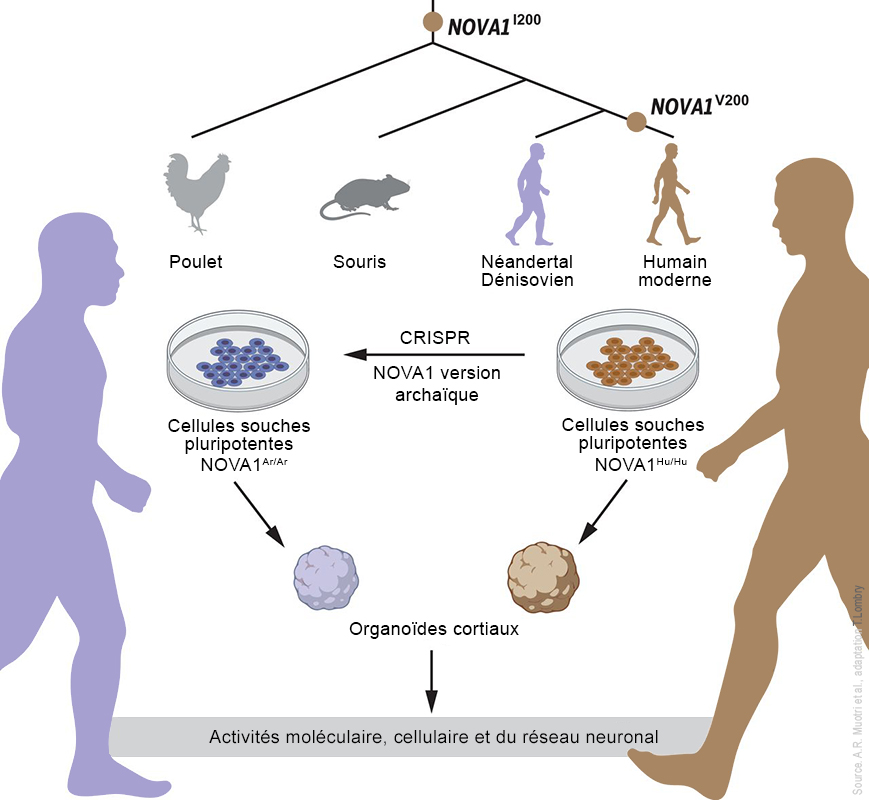
Les chercheurs ont découvert un haplotype protecteur d'environ 75 kb dérivé des Néandertaliens dans la région chromosomique 12q24.13. Cet haplotype contient une variante d'épissage (la phase de coupure et d'élimination des extrons permettant la ligature des seuls introns permettant de former l'ARNm à partir de l'ADN) d'OAS1 qui n'existe que chez les personnes d'ascendance africaine, indépendamment du flux génétique des Néandertaliens. Selon les chercheurs, "Cette variante d'épissage est susceptible d'être le SNP responsable de l'association à ce locus, impliquant ainsi fortement que OAS1 est le gène effecteur influençant la gravité de la Covid-19." Pour rappel, les SNP ou "snips" (polymorphismes mononucléotidiques) sont le type de variation génétique le plus courant chez les humains. Chaque SNP représente une différence dans un seul nucléotide (par ex. un SNP peut remplacer la cytosine (C) par la thymine (T) dans un certain segment d'ADN). L'étude a montré que 80% des personnes d'ascendance africaine étaient porteuses de cette variante protectrice du gène (l'allèle rs10774671 G) qui détermine la longueur de la protéine codée par le gène OAS1. Des études antérieures ont montré que c'est justement la variante la plus longue de la protéine qui découpe le plus efficacement l'ARN du SARS-CoV-2 et protège ces personnes contre l'hospitalisation liée au Covid-19. En revanche, l'allèle rs10774671 G est moins fréquent (32%) chez les individus d'ascendance européenne. Effets du variant du gène NOVA1 des humains archaïques Dans une étude publiée dans la revue "Science" en 2021, l'équipe d'Alysson Muotri de l'École de Médecine de l'Université de San Diego, en Californie (USDC), développa une plate-forme pour tester l'impact de trois variants génétiques humains archaïques dont NOVA1. Grâce à l'outil moléculaire CRISPR-Cas9, les chercheurs ont réintroduit la forme archaïque du gène NOVA1 trouvée chez les Néandertaliens et les Dénisoviens dans des organoïdes du cerveau humain (des cultures cellulaires souches pluripotentes reproduisant in vitro la structure micro-anatomique d'un cerveau). Ils ont ensuite mesuré son effet pendant le neurodéveloppement et constaté que les connexions neuronales étaient altérées. Selon les chercheurs, "La réintroduction de la version archaïque de NOVA1 dans un génome d'humain moderne provoque des changements d'épissage alternatif dans les gènes impliqués dans le développement neurologique, la prolifération et la connectivité synaptique. Ces changements sont accompagnés de différences de morphologie organoïde et de fonction du réseau neuronal, suggérant que ce gène joue un rôle fonctionnel. En outre, les organoïdes corticaux porteurs du gène archaïque NOVA1 ont présenté des changements synaptiques distincts qui peuvent avoir conduit aux altérations observées dans le développement du réseau neuronal."
Sur le plan biologique de la neurotransmission, les données suggèrent que l'expression du gène archaïque NOVA1 modifie les interactions protéiques synaptiques et affecte la signalisation glutamatergique (un neurotransmetteur) lors des connexions neuronales. Selon les chercheurs, "un sous-ensemble de changements génétiques peut ainsi expliquer les traits phénotypiques qui séparent notre espèce des Néandertaliens. Ces résultats suggèrent que la substitution spécifique de NOVA1 chez l'Homme moderne, qui s'est fixée après la divergence avec les Néandertaliens, peut avoir eu des conséquences fonctionnelles sur l'évolution de notre espèce." Si la pathologie observée explique en partie les différences cérébrales entre les deux espèces humaines, elle permettrait aussi aux chercheurs de mieux comprendre certaines maladies neurologiques dont nous avons peut-être héritées dans un lointain passé, telles que l'autisme qu'étudie également l'équipe de Muotri. Pour ceux qui doutaient encore de cet héritage génétique ou de ses conséquences, en voici un effet inattendu. Deuxième partie Les hybridations chez les Dénisoviens
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||