|
|
|
Origine et évolution de l'Homo sapiens
Vers une révision de l'évolution des Homo sapiens (II) 4. La nécessité d'une vision plus complexe Exposons à présent les raisons pour lesquelles les modèles génétiques doivent intégrer une vision plus complexe de l'ancienne migration et de la divergence des populations africaines. Le point de départ de la plupart des études génétiques sur les origines humaines a été d'étudier en profondeur la diversité actuelle entre et au sein des populations africaines. La plupart des études ont utilisé de simples modèles démographiques "arborescents" pour déduire les époques de divergence, en négligeant ou en simplifiant la stucture des populations, même si l'on considère parfois un certain degré de flux génétique entre les embranchements (cf. Encadré 2). De telles études ont produit diverses estimations des bifurcations dont celle des populations Khoïsan (ou KhoeSan) vivant en Afrique australe et représentée par les ethnies non-bantoues San ou Bushmen (Bochimans) et leurs voisins les Khoïkhoïs ou Hottentots qui ont conservé les plus hauts niveaux de diversité génétique parmi les populations humaines actuelles, au point qu'elle possède encore la trace génétique d'une ancienne divergence inférée remontant entre 150000 et 300000 ans[52], [53], [54], [55], [56], [57], [58]. On y reviendra à propos de la sortie d'Afrique de l'Homo sapiens moderne. Certains auteurs ont interprété ces données en conjonction avec un gradient de diversité génétique africaine décroissant du sud vers le nord favorisant un modèle d'origine unique des humains modernes en Afrique australe plutôt qu'en Afrique de l'Est[59], [60]. La variabilité inférée observée dans les bifurcations reflètent la variété des méthodologies, des hypothèses modélisées et des sources de données, avec une tendance générale que les analyses plus récentes infèrent des dates plus anciennes. De plus, à l'ancien flux génétique et des structures, s'ajoute des mouvements plus récents de populations en Afrique tels que l'expansion de la langue bantoue chez les peuples de l'Afrique de l'Ouest il y a 2000 à 1500 ans[52], [61] qui peuvent cacher les signatures d'anciens processus démographiques, comme c'est le cas des épisodes migratoires de "retour vers l'Afrique" en provenance d'Europe et de l'Asie du Sud-Ouest observés dans plusieurs régions du continent[60], [62], [63], [64], [65]. A consulter : Glossaire
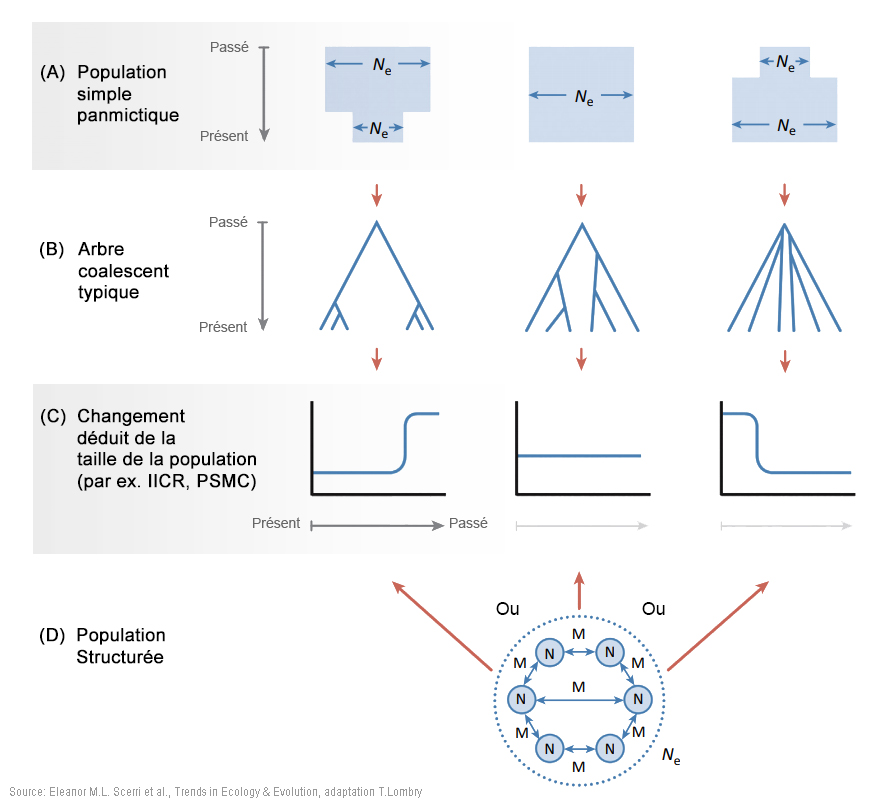
Les modèles incorporant une structure de population plus complexe peuvent être considérablement plus riches en paramètres et donc plus difficiles à tester par ordinateur, en particulier avec des données limitées. Cependant, ils offrent une vue plus générale et plus flexible de la démographie passée - dont on peut se contenter, mais sans s'y limiter - que les modèles arborescents de population plus traditionnels. En outre, la disponibilité d'un nombre croissant de données génomiques et les développements dans les méthodes analytiques autorisent des inférences dans des modèles plus complexes et plus réalistes. Ces développements ont montré que la structure ne peut pas être négligée et peut générer des schémas dans les données génétiques similaires à ceux générés par d'autres formes de changement démographique (par exemple [66], [67]). Ainsi, inférer les changements dans la taille effective de la population (Ne) peut entraîner des changements dans les relations entre les anciennes populations plutôt qu'à partir de, ou en plus des changements dans la taille de la population échantillonnée[7], [68]. En effet, la relation entre la taille inférée de Ne et la taille de la population échantillonnée n'est pas évidente et peut même être contre-intuitive quand une structure existe[7], [66], [68]. L'échelle géographique à laquelle la structure génétique de la population a pu exister est également difficile à déduire. Par exemple, une étude génomique récente a montré l'existence d'une structure entre les populations humaines pré-agricoles séparées de seulement quelques dizaines ou centaines de kilomètres[69]. Ces conclusions défient voire vont contre l'idée que le début de la préhistoire de notre espèce peut être correctement approchée à partir de la croissance de la population au sein d'une seule lignée[70]. Bien que les données génomiques modernes aient été forgées par leur passé démographique et contiennent de ce fait de grandes quantités d'informations, ces données peuvent être expliquées par de nombreux modèles différents d'histoires de la population, ce qu'on appelle l'équifinalité (malgré différents points de départ, les processus vivants atteignent le même état final). De plus, tous ces modèles sont nécessairement des abstractions et des simplifications des véritables histoires de la population et les divergences constatées peuvent être particulièrement problématiques pour résoudre certaines questions relatives au passé (spécifications erronnées). Cela signifie que la structure peut être difficile à détecter sans ambiguïté et encore plus difficile à reconstruire. Ainsi, plusieurs études sur des populations africaines ont identifié des gènes ayant des temps de coalescence de l'ordre de 1 million d'années, ce qui pourrait être interprété comme indiquant un métissage avec des homininés archaïques[9], [10], [73]. Cependant, même dans une seule population humaine, on s'attend à de très anciens temps de coalescence pouvant dépasser 1 million d'années (cf. Figure 1 ci-dessous à gauche). Par conséquent, des inférences basées sur la queue de la distribution des temps de coalescence, qui sont particulièrement sensibles à une spécification incorrecte du modèle, doivent être interprétées avec prudence. En effet, plusieurs auteurs ont soutenu que des coalescences très anciennes sont compatibles avec une seule lignée humaine originaire d'Afrique avec une structure de la population très profonde[7], [68], [74]. Les données d'anciens ADN (aADN) datant du Pléistocène peuvent fournir un moyen de résolution supplémentaire et des études sur des individus de l'Holocène ont récemment révélé une structure extensive et l'activité migratoire au cours de cette période[8], [75]. L'aADN du Pléistocène serait plus instructif mais il est difficile à obtenir du fait que les environnements tropicaux (humides) sont généralement défavorables à la préservation de l'ADN. Cependant, une étude récente a montré que l'aADN du Pléistocène supérieur peut être récupéré dans certaines régions africaines[65]. Ces études démontrent que les déductions faites à partir des schémas de la diversité génomique humaine doivent prendre en compte la fluctuation de la structure de la population sur de longues périodes, en complément des divers modèles de l'origine de la population panmictique africaine. 5. Facteurs environnementaux et écologiques de la structure de la population Les données génétiques, fossiles et archéologiques discutées ci-dessus indiquent que l'Homo sapiens a évolué dans des populations très structurées, probablement dans de nombreuses régions d'Afrique. Elucider le degré et les mécanismes des structures sous-jacentes de la population nécessitera de tenir compte de la variabilité environnementale et donc de l'évolution du climat au Pléistocène supérieur dans l'espace et dans le temps (par exemple [13] et Figure 2 ci-dessous au centre et à droite). Des études ont montré que des refuges glaciaires furent des catalyseurs clés du changement évolutionniste[76] et auraient certainement généré la structure de la population. Néanmoins, certaines régions formant des lagunes et des îles habitées peuvent également avoir joué un rôle central dans la survie des populations reliques.
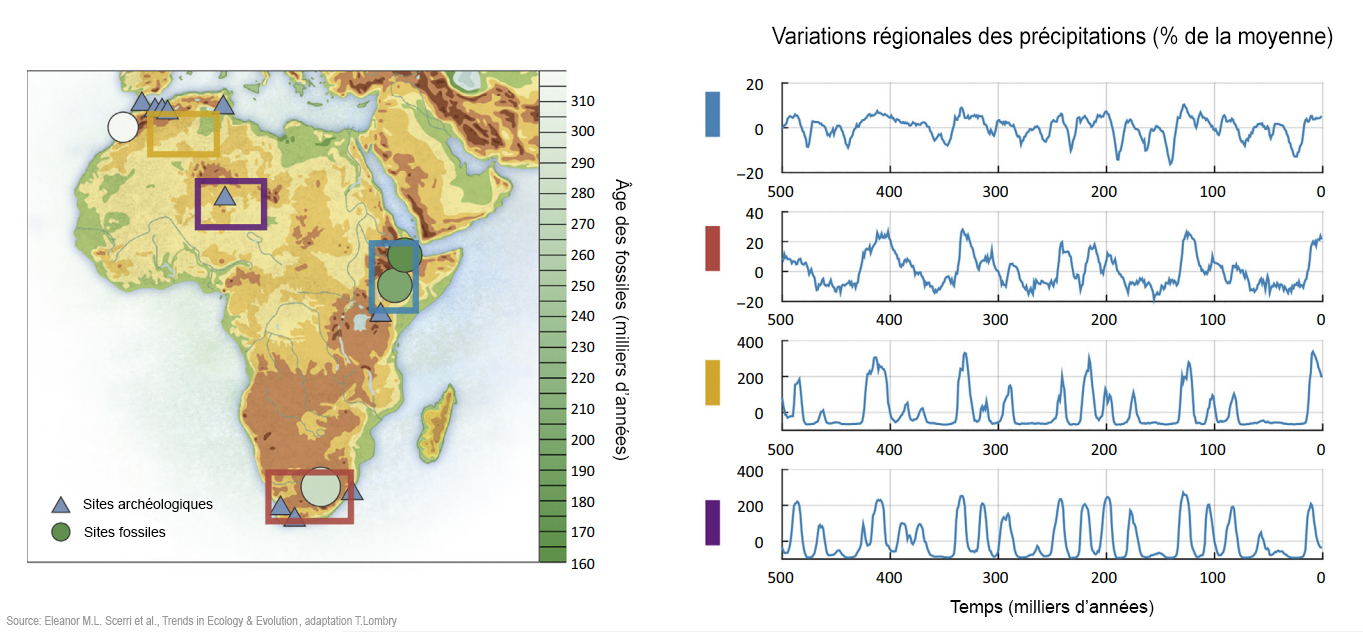
Sur le plan climatique, les recherches ont mis en évidence de vastes changements environnementaux asynchrones dans différentes régions d'Afrique (par exemple [13], [77]. Les extrémités nord et sud du continent sont les plus fortement affectées par les précipitations hivernales d'ouest dont la variabilité dépend largement des changements des conditions dans l'océan Atlantique. Cependant, la majeure partie de l'Afrique connaît des pluies de mousson dépendantes de la zone de convergence intertropicale (ITCZ) dont la force et l'emplacement varient selon les changements de l'insolation qui sont principalement influencés par les cycles de Milankovitch dont la précession de l'axe de rotation de la Terre. Par conséquent, les parties de l'Afrique tropicale qui sont actuellement humides connurent pas le passé de nombreux épisodes d'aridité extrême[78], [79]. En même temps que la mousson se déplaça vers le nord, le Sahara s'est contracté et les réseaux de lacs et de rivières se sont étendus à une grande partie de l'Afrique du Nord[80], [81], [82] avec des conditions similaires dans certaines parties du sud-ouest de l'Asie. Des changements à petite échelle ont également été observés dans la mousson. Par exemple, en Afrique de l'Ouest, la savane et les zones boisées sont fortement affectée par de petits changements dans le régime des précipitations[83], [84]. Le climat variait donc considérablement avec des périodes d'aridité ou d'humidité relativement croissantes et asynchrones à travers l'Afrique. Fondamentalement, ces facteurs sont les principaux moteurs de la structure de la population faunique et de la spéciation[85], [86], illustrés par de nombreux modèles phylogénétiques similaires de la distribution des animaux subsahariens. Par exemple, Bertola et ses collègues[87] ont montré que des dizaines d'espèces présentent des populations distinctes dans les deux principaux royaumes évolutionnaires de l'Afrique occidentale/centrale et de l'Afrique orientale/australe. Beaucoup d'espèces montrent également une subdivision supplémentaire entre l'Afrique de l'Est et l'Afrique australe, signifiant qu'il existait des refuges importants dans ces trois régions. Ces espèces occupent divers niveaux trophiques (les différentes positions dans une chaîne alimentaire), ce qui suggère d'un point de vue écologique que le climat affecta l'ensemble des communautés. Par conséquent, la spéciation faunique semble avoir été catalysée par un habitat climatiquement fragmenté et des interactions entre différents biomes au cours du temps. Cela donne un aperçu de la manière dont la structure de la population humaine aurait pu se maintenir sur des échelles de temps significatives et dans différentes zones géographiques de l'Afrique. En Afrique, le concept de "réseaux refuges" a été spécifiquement impliqué dans les subdivisions et les expansions de la population humaine du Pléistocène[18] et par conséquent ces régions ont un intérêt évolutif majeur. Bien qu'il ait été démontré que la fragmentation d'un habitat propice est un moteur majeur de la structure de la population après l'isolement par la distance (Encadré 1), l'isolement par l'habitat peut aussi jouer un rôle important chez l'animal[84] et probablement aussi dans la structure de la population humaine[14], [88]. Des défis majeurs subsistent quand il faut intégrer des preuves fossiles, archéologiques et des lignées génétiques dans des contextes paléoenvironnementaux et paléoécologiques (voir [3] et [72] pour quelques tentatives). Actuellement, la chronologie d'une grande partie du dossier paléoanthropologique reste trop grossière pour permettre de tirer des conclusions définitives sur le rôle des changements environnementaux. De nouvelles pistes de recherche prometteuses comprennent des analyses génomiques de la faune, y compris l'identification des espèces commensales (vivant en symbiose mais dont l'association ne produit aucun effet bénéfique ou préjudiciable sur l'organisme hôte) et des reconstructions d'habitats humains à partir des analyses d'isotopes stables. 6. Remarques finales : avancer Les données morphologiques, archéologiques, génétiques et paléoenvironnementales disponibles indiquent que la subdivision des populations humaines africaines du Pléistocène moyen et supérieur fut le moteur de l'émergence quasi-mosaïque et de l'évolution de la morphologie de l'Homo sapiens. Sur le plan reproductif, des populations semi-isolées se sont adaptées aux écologies locales parallèlement à la dérive génétique. Cet isolement de la population était probablement facilité par la taille réduite des populations. Ainsi, comme pour les autres animaux, on ne devrait pas considérer que le flux génétique fut constant au cours du temps ou se serait déroulé au même rythme dans et entre différentes régions. À travers les grandes échelles temporelles du Pléistocène moyen et supérieur, en raison des fortes fluctuations climatiques, le nombre de populations dans les zones intermédiaires reliant les différentes régions est également susceptible d'avoir considérablement fluctué. Plusieurs questions majeures toujours ouvertes découlent de cette réorientation des origines humaines récentes. Est-ce que les principales caractéristiques morphologiques diagnostiquées ont émergé dans une région et se sont élaborées au cours des dispersions ultérieures ? Ou est-ce que la transition de l'homme "archaïque" vers l'homme "moderne" - qu'elle soit visible dans sa morphologie ou dans la culture lithique - s'est produite progressivement et dans un mode mosaïque à travers le continent ? Si tel était le cas, l'hybridation archaïque africaine joue-t-elle aussi un rôle ? Comment les preuves existantes concernant la structure d'une population peuvent-elles améliorer notre compréhension de l'histoire des changements survenus dans la taille de la population et sa dispersion ? De même, nous n'avons pas une bonne compréhension de la concordance qui pourrait exister entre les structurations morphologique et culturelle. Les signatures culturelles régionales sont apparentes, ce qui soulève la possibilité que des formes spatialement distinctes de culture lithiques reflètent des schémas similaires d'isolement et d'agrégation de la population. Combler ces lacunes intellectuelles nous oblige à reconsidérer les concepts d'espèces paléoanthropologiques qui sont contestés par l'idée qu'il existerait une structure profonde de la population avec un flux ou mélange génétique sporadique. Finalement, reconstruire l'histoire démographique des populations humaines dans toute sa complexité est au-delà du pouvoir de la génomique des populations, nécessitant une approche interdisciplinaire. Dans le passé cet objectif fut atteint par des généticiens travaillant avec des archéologues et des paléoanthropologues qui sont parvenus à définir un ensemble restreint d'hypothèses simplifiées dont les résultats génétiques peuvent encore être utilisés pour identifier les modèles correspondant le mieux aux données factuelles. Alors qu'une telle approche a rencontré un succès considérable, un portrait plus complet exigera l'intégration de différents types de données (génétique, fossile, culture lithique, données paléoclimatiques et paléoécologiques) en utilisant les mêmes modèles analogues de la structure de la population, de son changement de taille et de sa dispersion. Cela représente un défi majeur pour l'inférence démographique ancestrale des prochaines années. La caractérisation complète de la nature de cet apparent "multirégionalisme africain" exige également de rejeter de nombreuses hypothèses implicites datant quelque peu et de formuler de nouvelles questions. Par exemple, le décalage chronologique entre les estimations génétiques temporelles des divergences morphologiques des populations et les changements dans les archives fossiles ne sont pas bien compris; on estime qu'ils ne devraient pas être courts - en particulier parce que les déductions tirées de données génétiques sont profondément influencées par les modèles ou les familles de modèles utilisés. Par exemple, les estimations des bifurcations temporelles dans une population peuvent s'avérer moins appropriées ou pertinentes dans le cadre de notre compréhension de l'évolution humaine lorsqu'on utilise des modèles tenant compte de la structure spatiale. De même, alors qu'un crâne globulaire d'Homo sapiens actuel semble représenter une synapomorphie (un caractère dérivé ou apomorphique partagé par deux ou plusieurs taxons-frères), pent-on le caractériser efficacement si nous l'appliquons aux enregistrements fossiles ? Nous soulignons que l'Homo sapiens est une lignée ayant des racines africaines profondes et probablement diverses qui remettent en question notre utilisation de termes tels que "Homo sapiens archaïque" et "humains anatomiquement modernes". À moins de les rendre opérationnelles grâce à des traits plus clairement définis, ces catégories auront une valeur décroissante. Les conclusions (diagnostics) concernant l'Homo sapiens doivent refléter des trajectoires d'évolution plutôt que des vues statiques de notre espèce - qui continue d'évoluer à différentes échelles. Les orientations de la recherche durant la prochaine décennie seront cruciales pour résoudre ces nouveaux thèmes de recherche. Les génomes humains contemporains sont aujourd'hui disponibles pour l'ensemble du continent africain avec un nombre croissant de génomes anciens. Notre compréhension de la paléoécologie s'est également améliorée grâce à des reconstructions biogéographiques basées sur les génomes de la faune africaine. Les reconstructions paléoclimatiques sont de plus en plus précises, avec un plus grand nombre de données de proxies et de meilleurs modèles couvrant les périodes clés. Enfin, l'expansion des investigations paléoanthropologiques dans les zones négligées de l'Afrique révélera sans aucun doute de nouvelles données qui vont considérablement affiner les paramètres de l'évolution humaine récente. Plus plus d'informations L'origine et l'avenir de l'homme (les premiers Homo sapiens) Homo sapiens, un taxon polytypique ?
|
|||||||||||||||||||||


{kind=link}