|
|
|
Microbiologie du Covid-19
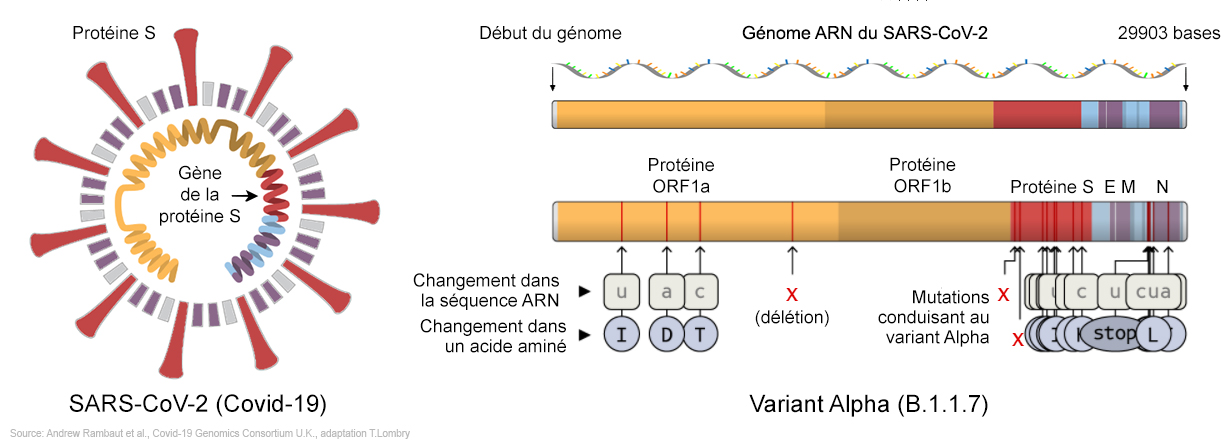
Les différents variants (III) Pour rappel, les mots "variant" et "souche" sont synonymes. Seule différence, le mot "variant" est plus moderne car il insiste sur les mutations, les variations génétiques du virus. Insistons sur le fait que l'apparition de variants n'étonne pas les chercheurs. Comme nous l'avons expliqué, le SARS-CoV-2 évolue et mute constamment, comme le font tous les virus. De tels changements sont donc prévisibles. Un problème se pose lorsqu'un variant présente une mutation qui facilite sa propagation ou lui permet d'échapper aux défenses du système immunitaire. On y reviendra. Premières mutations La première mutation du SARS-CoV-2 fut observée le 8 janvier 2020 chez un patient de Wuhan (génome WH-09). Il s'agit de la substitution de la base nucléique C par une base nucléique U sur la 186e position de son ARN. En dehors de Wuhan, la même mutation fut trouvée le 27 février 2020 sur un autre patient à 1000 km de là, à Guangzhou (Canton). Cet échantillon (génome GZMU0030) pourrait être un descendant direct de celui de Wuhan ou il peut s'agir d'un autre variant partageant un ancêtre commun. Au cours des semaines suivantes, le variant de Guangzhou subit deux autres mutations non silencieuses : la première sur la protéine non structurale ORF1a où un acide aminé muta en isoleucine (I), la seconde sur la protéine E où une acide aminé fut également modifié. Arrêtons-nous un instant pour souligner que ces changements génétiques concernent la mutation d'une seule base nucléique sur ORF1 par exemple qui comprend environ 20000 bases. Son effet sur la fonction du gène qu'elle affecte (cf. les biostatistiques page précédente) est aussi insignifiant que remplacer la couleur d'une seule pièce d'un puzzle Comme expliqué précédemment, le virus peut ainsi subir des centaines de substitutions de ses acides aminés sans pour autant altérer la manière dont ses protéines clés s'attachent aux cellules qu'il infecte. C'est sur ce principe que sont développés les vaccins qui restent efficaces malgré les nombreuses mutations du virus qui surviennent entre leur développement et leur mise sur le marché. Mais à l'impossible, nul n'est tenu. Il arrive un jour où le nombre de mutations est tel que le vaccin n'est plus adapté au virus et perd son efficacité. Il est alors grand temps de développer un nouveau vaccin adapté au nouveau variant, ce qui peut parfois se réaliser en moins d'un mois. Ensuite, lorsque le virus se propagea dans toute la Chine puis sur les autres continents, des variants spécifiques se développèrent dans certaines régions ou villes (Shanghaï, Beijing, Lombardie, Haute-Savoie, New York, Washington, Californie, Victoria en Australie, etc). Parmi ces variants, le génome BavPat1 découvert à Munich provenait de Wuhan via Shanghaï, un génome quasi identique au BJ2460 de Beijing fut découvert en Belgique, le génome WA1 de Seattle et UC4 de Californie provenaient de personnes ayant visité Wuhan. D'autres variants comme ceux de New York avaient transité par l'Iran (génome NY1-PV08001) ou par l'Europe. Enfin, les variants européens et américains sont devenus de nouveaux foyers infectieux qui exportèrent le virus en Amérique du Sud et le réinduisirent en Asie.
Au fil des mois, certaines parties du génome du SARS-CoV-2 subirent de nombreuses mutations. Certaines protéines furent peu modifiées et d'autres pas du tout. Entre décembre 2019 et avril 2020, quelque 4400 acides aminés du génome du SARS-CoV-2 furent substitués, certains emplacements du génome qu'on appelle des "hot spots" étant plus tolérants aux mutations, d'autres ne tolérant aucune mutation et étant critique pour le virus. 185 mutations sont apparues de façon récurrente. Au total, en 2020 les virologues ont recensé plus de 12000 mutations du SARS-CoV-2. Si c'est impressionnant, en les analysant on constate que la majorité d'entre elles n'a pas amélioré sa transmissibilité (cf. L.von dorps et al., 2020). En revanche, les nouveaux variants découverts en fin d'année sont plus transmissibles (voir plus bas). Nous avons constaté que le SARS-CoV-2 fut particulièrement meurtrier en Italie, au Brésil et à New-York. En Italie, il y eut 919 décès le 27 mars 2020, 615 décès au Brésil le 6 mai 2020 et 574 décès rien qu'à New York le 4 avril 2020 (et 2624 décès aux Etats-Unis le 21 avril 2020). Si la raison de cette hécatombe très localisée n'est pas encore établie, quelques pistes ont été explorées. Même si elles ne sont pas encore convaincantes, elles suggèrent qu'il existerait des variants très virulents (sans écarter les facteurs de risque ainsi que les prédispositions génétiques et/ou immunitaires).
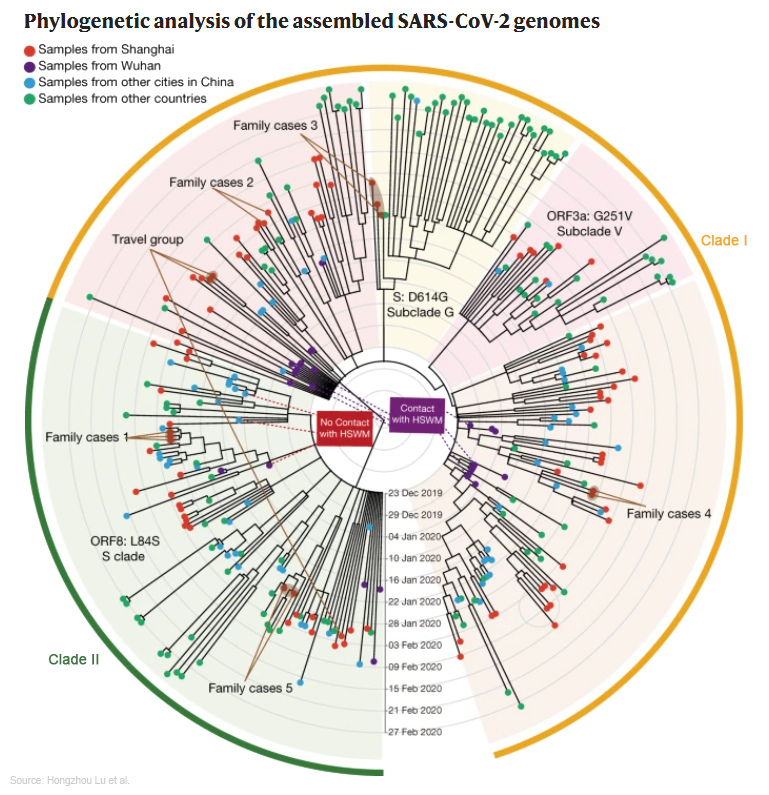
Dans une étude publiée dans la revue "National Science Review" le 3 mars 2020, Jian Lu et ses collègues ont analysé 103 génomes complets prélevés sur des patients chinois, australiens et coréens et sont arrivés à la conclusion qu'il existerait deux variants ou souches distinctes du SARS-CoV-2 : "la souche L et la souche S". La souche L aurait muté à partir de la souche S et serait plus agressive puisqu'elle serait responsable de 70% des contaminations contre 30% pour la souche S. Une autre étude réalisée sur 11 patients comprenant 8 hommes et 3 femmes publiée en avril 2020 suggère également l'existence de plusieurs variants dont certains présentent une charge virale 270 fois plus importante que d'autres. Certains variants ont subi 33 mutations dont 19 sont nouvelles. Sur base des analyses faites par GISAID, les chercheurs concluaient que les variants du SARS-CoV-2 circulant à l'époque en Europe et à New York seraient les plus virulents tandis que ceux circulant ailleurs dans les États-Unis seraient les plus modérées (cf. M.Zheng et al., 2020). Mais cette hypothèse n'a pas été confirmée. En revanche, nous verrons que certains variants sont plus transmissibles que d'autres. Mais ce n'est peut-être pas la seule raison qui les rend plus virulents. Enfin, la sensibilité de la personne contaminée (ses gènes, son âge, son genre, ses facteurs de risque, etc.) représente aussi un facteur déterminant. Comme l'étude précédente de l'équipe de Tang, les conclusions de Zheng et ses collègues furent également critiquées compte tenu du très faible échantillonnage et du séquençage simple (chaque gène n'est lu qu'une fois) introduisant potentiellement un taux élevé d'erreurs. Néanmoins, cette hypothèse confirme une étude de l'Université Northeastern qui avait déjà montré que le variant du SARS-CoV-2 présente à New York provenait d'Europe, probablement via l'Italie, où il fit énormement de victimes. Dans une autre étude publiée dans la revue "Nature" le 20 mai 2020, Hongzhou Lu de l'Université Fudan de Shanghaï et ses collègues ont analysé les données cliniques, moléculaires et immunologiques de 326 patients Covid hospitalisés à Shanghaï. Comme on le voit sur le cladogramme présenté à gauche, les séquences génomiques virales de 94 patients Covid furent analysées et ont montré une évolution stable des variants. Dans l'échantillonnage, il y avait deux lignées majeures (Clade I et II) avec un historique d'exposition différentiel pendant la phase précoce de l'épidémie à Wuhan. Néanmoins, ils ont montré une virulence et des résultats cliniques similaires, en particulier dans la progression de la maladie. Selon les chercheurs, "Les déterminants de la gravité de la maladie semblaient provenir principalement de facteurs de l'hôte tels que l'âge et la lymphocytopénie (et sa tempête de cytokines associée), alors que la variation génétique virale n'affectait pas de manière significative les résultats". Ceci est une très bonne nouvelle pour le développement d'un vaccin. Selon les données du CDC américain, de la base Nextstrain et de l'OMS, voici les caractéristiques des principaux variants du SARS-CoV-2. VOC et VOI Pour les chercheurs, il existe plusieurs types de variants, les VOC (variants of concern ou variants préoccupants), les VOI (variants of interest ou variant d'intérêt) et les autres variants sous surveillance. Ces acronymes sont parfois utilisés dans les articles scientifiques. Voici les définitions de l'ECDC.
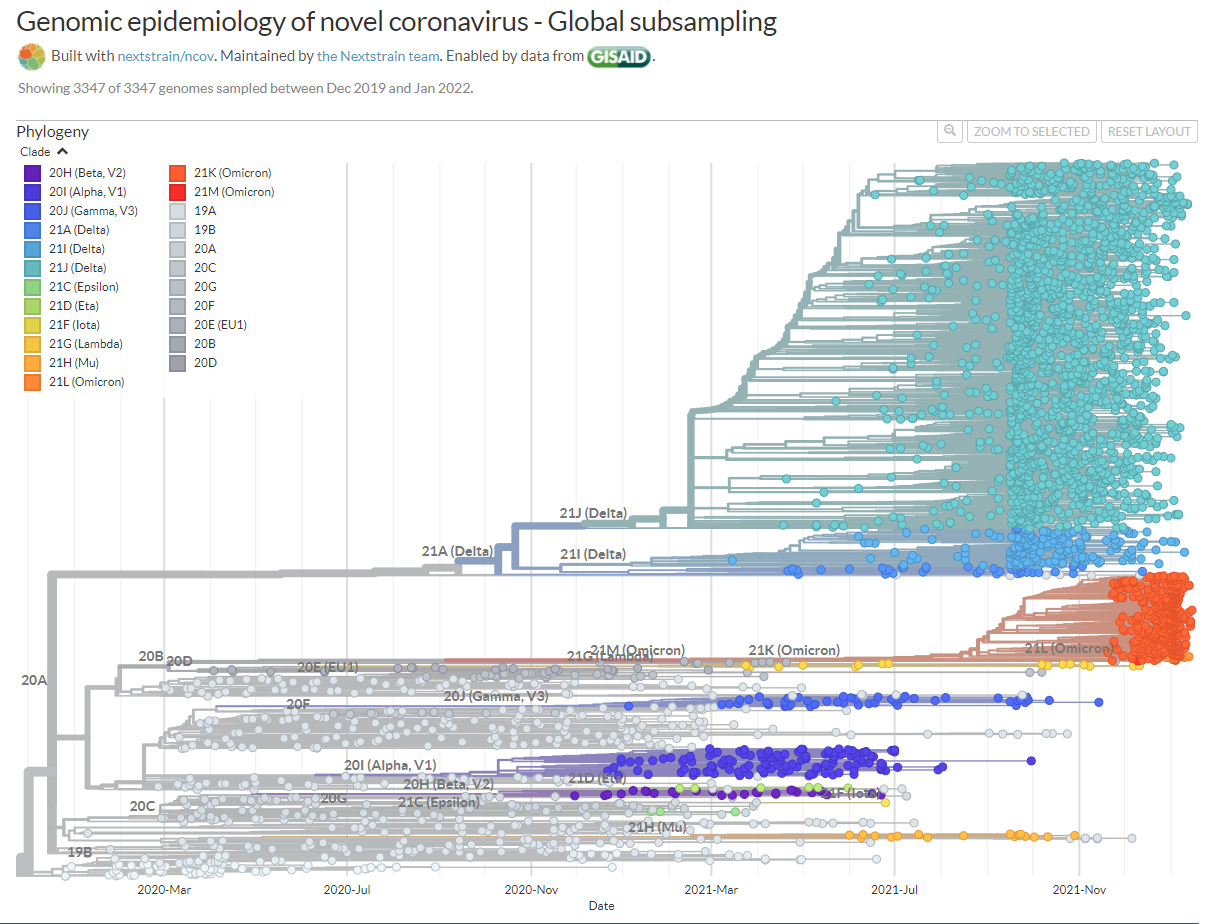
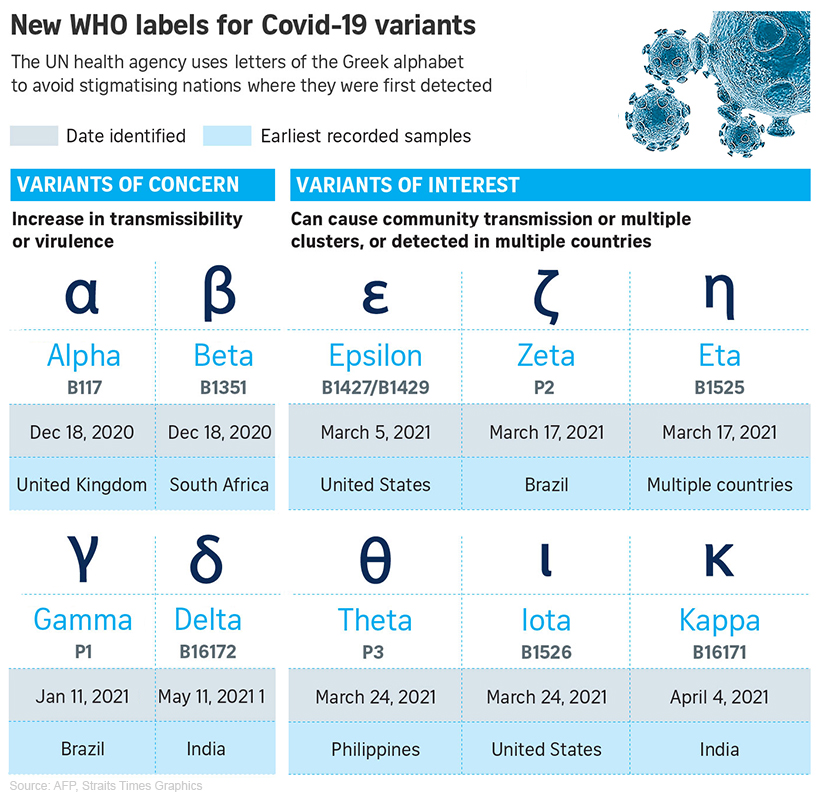
Les VOC : pour ces variants, des preuves claires sont disponibles indiquant un impact significatif sur la transmissibilité, la gravité et/ou l'immunité qui est susceptible d'avoir un impact sur la situation épidémiologique. Les preuves génomiques, épidémiologiques et in vitro combinées de ces propriétés invoquent une confiance au moins modérée. En outre, tous les critères pour les variants d'intérêt et sous surveillance décrits ci-dessous s'appliquent. Les VOC du SARS-CoV-2 sont : B.1.1.7, B.1.1.7+E484K, B.1.351, P1, B.1.617.2, et B.1.716.2.1. Les VOI : pour ces variants, des preuves sont disponibles sur les propriétés génomiques, des preuves épidémiologiques ou des preuves in vitro qui pourraient indiquer un effet significatif sur la transmissibilité, la gravité et/ou l'immunité, ayant un impact réaliste sur la situation épidémiologique. Cependant, les preuves sont encore préliminaires ou associées à une incertitude majeure. En outre, tous les critères pour les variants sous surveillance décrits ci-dessous s'appliquent. Les VOI du SARS-CoV-2 sont : B.1.525, B.1.427/B.1.429, P.3, B.1.616, B.1.617.1, B.1.617.3, B.1.620, B.1.621, C.37 et C.1.2. Les autres variants sous surveillance. Ces variants supplémentaires du SARS-CoV-2 ont été détectés grâce à des systèmes d'intelligence artificielle épidémique, au dépistage de variants génomiques basé sur des règles ou sur des preuves scientifiques préliminaires. Il y a des indications qu'ils pourraient présenter des propriétés similaires à celles d'un VOC, mais les preuves sont faibles ou n'ont pas encore été évaluées par l'ECDC (ou le CDC). Les variants énumérés ci-dessous doivent être présents dans au moins une épidémie, détectés dans une communauté en Europe, ou il doit y avoir des preuves qu'il existe une transmission communautaire du variant ailleurs dans le monde. Ces autres variants sont : B.1.214.2, A.23.1+E484K, A.27, A.28, C.16, B.1.351+P384L, B.1.351+E516Q, B.1.1.7+L452R, B.1.1.7+S494P, C.36+L452R, AT.1, B.1.526, B.1.526.1, B.1.526.2, B.1.1.318, P.2, B.1.1.519 et AV.1. Ce classement peut bien entendu évoluer au cours du temps. En résumé, lorsqu'un virus surveillé est reclassé de VOI à VOC, il présente davantage de risques pour le public. Comme le rappelle l'OMS, "Plus le SARS-CoV-2 circule, plus il a de possibilités de muter" et plus il faut enrayer rapidement sa propagation. A consulter : SARS-CoV-2 variants of concern, ECDC, May 2021 SARS-CoV-2 Variant Classifications and Definitions, CDC, May 2021 Overview of Variants/Mutations, CoVariants Spike variants (liste des mutations du SARS-CoV-2), U.Stanford Nouvelle nomenclature de l'OMS Le 31 mai 2021, l'OMS proposa un nouveau système de codage pour nommer les variants du SARS-CoV-2. Jusqu'alors un variant pouvait s'appeller par exemple B.1.1.7 ou 20I/501Y.V1 ou encore VOC 202012/01. Certains pays hésitaient à déclarer un nouveau variant car dans certains cas il prend le nom du pays. Pour ne pas stigmatiser certaines pays et afin de rendre la nomenclature neutre, l'OMS décida d'utiliser les lettres de l'alphabet grec : Alpha, Beta, Gamma, etc. Cette nouvelle nomenclature a été appliquée aux bases de données GISAID, Nextstrain et Pango. Nous verrons ce que décidera l'OMS quand elle aura atteint la 24e et derrière lettre de l'alphabet, Omega.
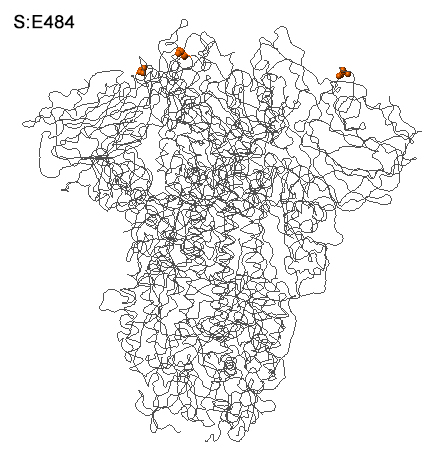
Document publié le 1 juin 2021 adapté de AFP/The Straits Times. Suivant cette nomenclature, les variants du SARS-CoV-2 les plus actifs sont : Alpha (B.1.1.7 et Q), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2 et AY), Epsilon (B.1.427 et B.1.429), Zeta (P.2), Eta (B.1.525), Theta (P.3), Iota (B.1.526), Kappa (B.1.617), Lambda (C.37) et Mu (B.1.621). Avec le temps, certains variants sont devenus dominants avant de céder la place à une autre mutation encore plus transmissible. Selon le CDC, aux États-Unis, en date du 4 septembre 2021, il y avait plus de 200 variants actifs. Ce variant mal nommé est en fait la mutation S:E484 dans le domaine de liaison au récepteur (RBD). Elle mérite la préséance car c'est la seule mutation du SARS-CoV-2 découverte à ce jour capable de "contaminer des cellules respiratoires humaines ACE2-négatives" pour reprendre les termes des chercheurs. En deux mots, il peut pénétrer dans les cellules humaines, même dépourvues de récepteurs ACE2 qui est la porte d'entrée standard du virus. Cette découverte fut décrite dans les "Cell Reports" le 13 juillet 2021 par l'équipe de la microbiologiste Sebla B. Kutluay de l'Ecole de Médecine de l'Université Washington à St. Louis. Cette découverte est à la fois surprenante et inquiétante car les scientifiques cherchaient de nouveaux modèles de cultures cellulaires des poumons pour étudier le mode d'infection du virus chez les patients Covid quand ils découvrirent par hasard ce variant dans le récepteur H522 des cellules humaines d'adénocarcinome du poumon.
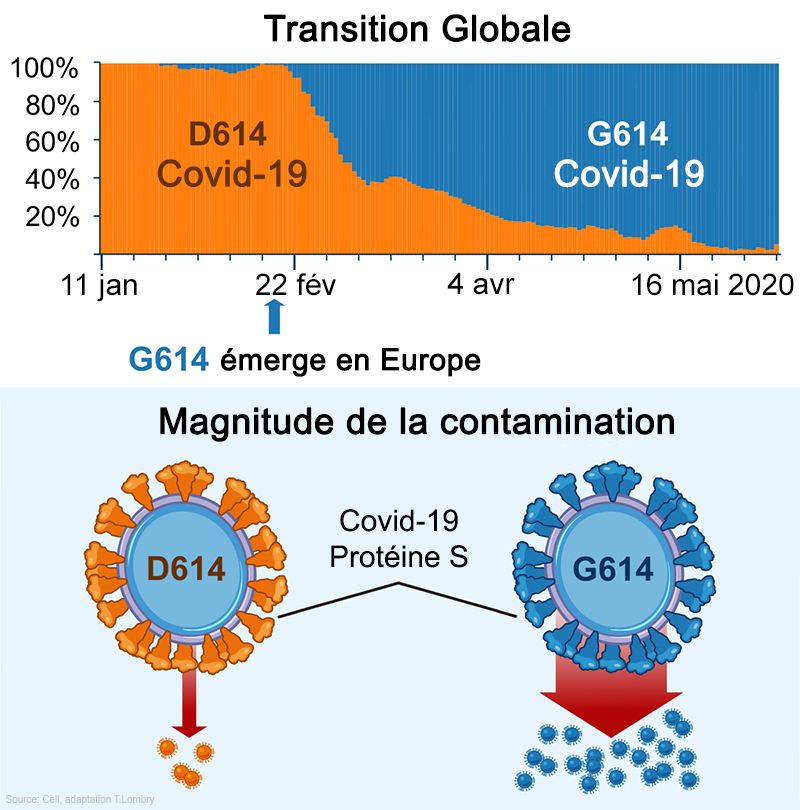
Ces cellules n'expriment pas l'ACE2 mais assurent tout de même la réplication du virus via sa protéine S comme illustré à droite. Selon les chercheurs, "la contamination de H522 par le SARS-CoV-2 entraîne des modifications du protéome dans les voies de réplication de l'IFN de type I, du cycle cellulaire et de l'ADN". Autrement dit le variant E484D modifie la dynamique des protéines impliquées dans la production des interférons qui jouent un rôle dans l'immunité innée et perturbent le cycle de vie des cellules. Ce variant est capable d'échapper aux défenses classiques du système immunitaire. Etant plus performant que les autres variants, il semble apparemment plus contagieux. De nouvelles études viendront compléter ce portrait préliminaire. Le variant D614G (EU) Dans un article publié dans la revue "Cell" le 2 juillet 2020 par la biologiste Bette Korber du Laboratoire National de Los Alamos (LANL) et ses collègues, les chercheurs ont constaté que le SARS-CoV-2 est devenu plus contagieux avec le temps. Le variant dominant à partir de mars 2020 nommé D614G (ou G614) provenait d'Europe et s'est ensuite installé aux Etats-Unis.
Dans une étude publiée un mois plus tôt sur "bioXrev" (non validée), les chercheurs avaient montré que ce variant ne diffère de son ancêtre que par une seule lettre de l'ARN caractérisant la protéine S. Dans ce premier article, Korber qui est spécialisée en biologie moléculaire et génétique des populations virales avait affirmé à propos de D614G que "lorsqu'il est introduit dans de nouvelles régions, il devient rapidement la forme dominante". Cette conclusion fut critiquée par leurs confrères car les chercheurs n'avaient pas prouvé que la mutation elle-même avait provoqué sa domination. En effet, c'est peut-être le résultat d'autres facteurs ou l'effet du hasard. Les chercheurs ont donc réalisé des analyses et des expériences complémentaires, notamment à la demande des éditeurs de la revue "Cell". Il ressort de cette étude que sur les 999 patients examinés, ceux porteurs de cette mutation virale produisaient plus de virions mais sans que cela ne change la gravité de leur maladie, ce qui est rassurant. Quant aux expériences en laboratoire, elles ont montré que la nouvelle mutation pouvait contaminer 3 à 6 fois plus plus de cellules humaines. Korber affirma que "Le variant du SARS-CoV-2 qui domine aujourd’hui dans le monde infecte plus facilement les cellules que celui qui est apparu à l'origine en Chine, ce qui le rend probablement plus contagieux entre humains bien que cela reste à confirmer". Interrogé le 2 juillet 2020 par un chroniqueur du journal "JAMA" (cf. la vidéo sur YouTube), l'immunologue Anthony Fauci, directeur de l'Institut des maladies infectieuses américain (NIAID) et responsable de la Task Force Covid-19 auprès de la Maison Blanche déclara : "Nous ne savons pas encore si une personne s'en sort moins bien ou non [avec le nouveau variant D614G]. Il semble que le virus se réplique mieux et puisse être plus transmissible, mais nous en sommes toujours au stade d’essayer de le confirmer. Mais il y a de très bons généticiens des virus qui travaillent là-dessus". En résumé, les chercheurs concluaient en juillet 2020 que si le variant D614G du Covid-19 est sans doute plus infectieux que le virus original de Wuhan, il n'est pas forcément plus transmissible entre humains. Mais deux nouvelles études ont corrigé cette dernière affirmation. Dans une étude publiée dans la revue "Nature" le 26 octobre 2020, soit en ayant plus de 8 mois de recul par rapport aux premières études sur le nouveau variant, le microbiologiste Kenneth S. Plante de la Branche Médicale de l'Université du Texas (UTMB) et ses collègues ont examiné la mutation subie par D614G (alias USA-WA1/2020) dans sa protéine S et caractérisé son effet.
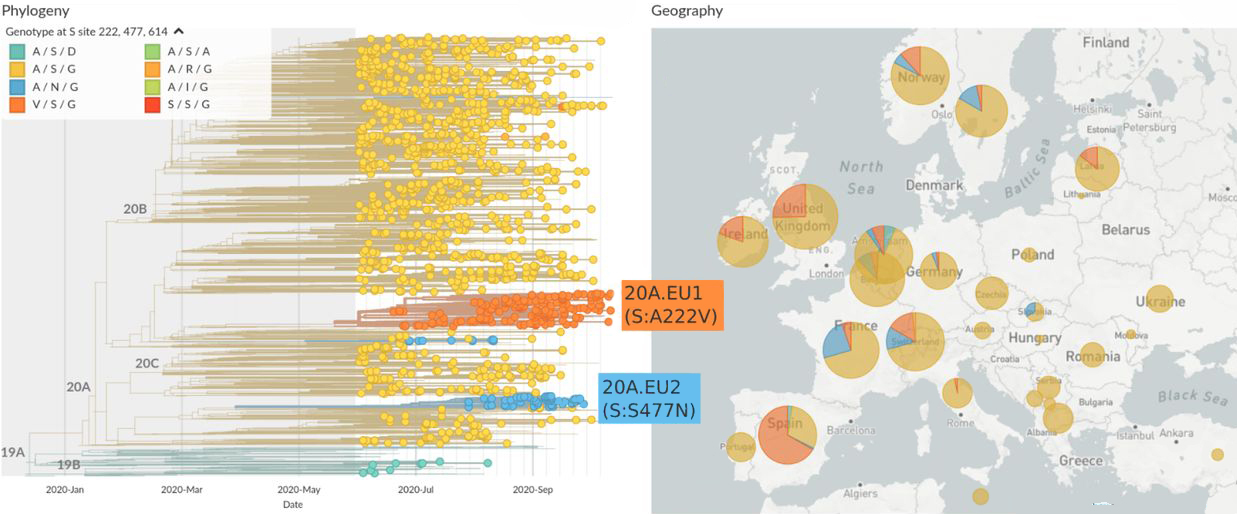
Selon les auteurs, le variant "D614G accroît la réplication sur les cellules épithéliales pulmonaires et les tissus des voies respiratoires humaines primaires grâce à une amélioration du pouvoir infectieux des virions". Les chercheurs ont infecté des hamsters par le variant G614 qui a produit des titres infectieux plus élevés dans les échantillons nasaux et de la trachée, mais pas dans les poumons, "confirmant les preuves cliniques que le variant D614G augmente les charges virales dans les voies respiratoires supérieures des patients Covid et peut augmenter la transmission". Ils ont également constaté que les sérums de hamsters infectés par le variant D614 présentent des titres de neutralisation légèrement plus élevés contre le variant G614 que contre le D614, indiquant que "la mutation ne réduit peut-être pas la capacité des vaccins dans les essais cliniques contre le Covid-19 et que les anticorps thérapeutiques doivent être testés contre le virus G614 en circulation". On y reviendra. Ils soulignent enfin l'importance du variant D614G dans la propagation de l'épidémie, l'efficacité des vaccins et de la thérapie par anticorps. Une autre étude publiée dans la revue "Science" le 12 novembre 2020 par Yoshihiro Kawaoka des universités du Wisconsin et de Tokyo et ses collègues confirme que le variant D614G présente une réplication ex vivo et une transmission in vivo plus efficaces (cf. le génie génétique) Selon les chercheurs, "Le variant D614G présente une infectiosité, une réplication et une aptitude compétitive plus efficaces dans les cellules épithéliales des voies respiratoires humaines primaires, mais conserve une morphologie et des propriétés de neutralisation in vitro similaires, par rapport au virus ancestral sauvage." En d'autres termes, le variant D614G du SARS-CoV-2 s'est adapté. Pour une charge virale similaire dans les tissus respiratoires, il infecte beaucoup plus rapidement les cellules. C'est notamment ce variant qui est l'origine de la perte d'odorat (anosmie). Les infectiologues sont d'avis que cela ne change rien dans les mesures de protection à respecter pour ralentir l'épidémie. Enfin, dans une étude publiée dans la revue "Science Advances" le 16 avril 2021 sur le variant D614G, des chercheurs confirment que la forme G est plus infectieuse in vitro et est associée à une augmentation des charges virales dans les voies respiratoires supérieures. Des simulations montrent que l'infectiosité accrue de la forme G est probablement due à un taux plus élevé de liaisons réussies avec le récepteur ACE2 des cellules hôtes. Nous verrons que c'est aussi son point faible. Les variants 20A.EU1 et 20A.EU2 (EU) Dans un article publié sur "medRxiv" (non validée) le 28 octobre 2020, Emma B. Hodcroft de l'Université de Bâle en Suisse et ses collègues ont annoncé la découverte d'un nouveau variant du SARS-CoV-2 au début de l'été 2020, vraisemblablement en Espagne, qui s'est ensuite propagé à plusieurs pays européens. En juillet 2020, le variant nommée 20A.EU1 représentait plus de 40% des cas de contamination en Espagne. En dehors de l'Espagne, de fréquences très faibles avant le 15 juillet, elle passa en septembre à 40-70% en Suisse, en Irlande et au Royaume-Uni. Ce variant est également présent en Norvège, en Lettonie, aux Pays-Bas et en France. Elle s'est ensuite propagée en Nouvelle Zélande et à Hong Kong et probablement dans d'autres pays. Ce variant porte la mutation A222V sur la protéine S. Les tests montrent que les anticorps humains sont légèrement moins efficaces pour neutraliser les variants portant cette mutation. Un autre variant nommé 20A.EU2 est présent en France et en Slovaquie et émerge en Belgique, aux Pays-Bas, en Allemagne, en Norvège et en Suède. En novembre 2020, les chercheurs ne disposaient pas de suffisamment de données de séquençages pour évaluer leurs fréquences dans d'autres pays. Il est actuellement difficile de savoir si ces variants se propagent en raison d'un avantage de transmission du virus ou si une incidence élevée en Espagne suivie d'une diffusion par les touristes est suffisante pour expliquer l'augmentation rapide dans plusieurs pays.
Les chercheurs confirment que le variant 20A.EU1 serait apparu chez des travailleurs agricoles espagnols provenant de deux foyers connus situés dans le nord-est du pays. Le virus contamina ensuite des travailleurs en Aragon et en Catalogne avant de se propager dans la région de Valence et dans le reste du pays. Selon les chercheurs, "Le variant 20A.EU1 diffère des séquences ancestrales d'au moins 6 positions, dont la mutation A222V dans la protéine S et A220V dans la nucléoprotéine [...]. Le variant 20A.EU2 présente une mutation S447N sur la protéine S". Le variant Alpha (B.1.1.7, GB) L'existence du variant Alpha ou B.1.1.7, également connu sous les codes 20I/501Y.V1, VOC 202012/01 ou VUI-202012/01, fut annoncée par Matt Hancock, le Sécrétaire britannique de la Santé le 14 décembre 2020 et confirmée par Patrick Vallance, le conseiller scientifique du gouvernement britannique au cours d'une conférence de presse qui s'est tenue le 19 décembre 2020. Le variant Alpha fut identifié mi-septembre 2020 chez des patients hospitalisés à Londres et dans le Kent. Selon "The Guardian" qui rapporte un communiqué de l'agence Public Health England (PHE), au 13 décembre 2020 le nouveau variant avait contaminé 1108 personnes. Au 19 décembre 2020 il était à l'origine de 62% des contaminations enregistrées à Londres, de 43% dans le sud-est de l'Angleterre et de 59% dans l'est du pays et dominant à présent les autre variants (cf. ce tweet de Tony Cox). Le variant fut identifié aux États-Unis fin décembre 2020. Au départ, le variant Alpha avait surtout contaminé des jeunes de moins de 20 ans, mais au fil des semaines il apparaît qu'il contamine tout le monde sans aucune préférence d'âge. Vu sa propagation plus rapide et le nombre croissant de paients Covid aux soins intensifs depuis février 2021, ce variant est entre 35 et 90% plus contagieux ou transmissible que l'ancienne souche et semble aussi plus virulent car les personnes contaminées, jeunes ou adultes, peuvent présenter du jour au lendemain des symptômes graves exigeant une prise en charge aux soins intensifs. A consulter : Variants of interest and concern in the EU/EEA, ECDC
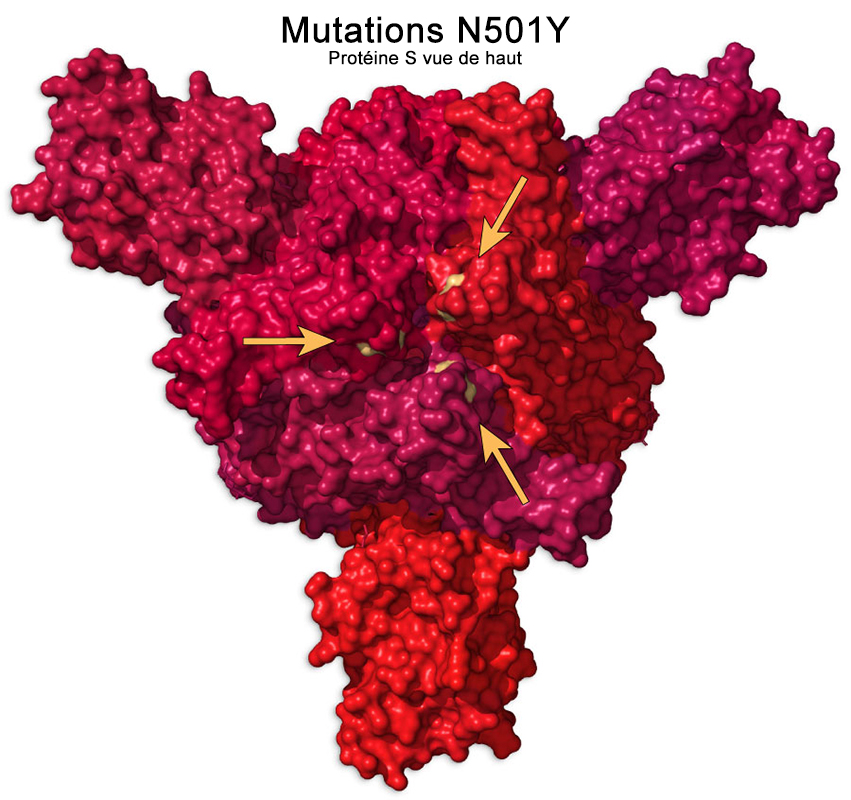
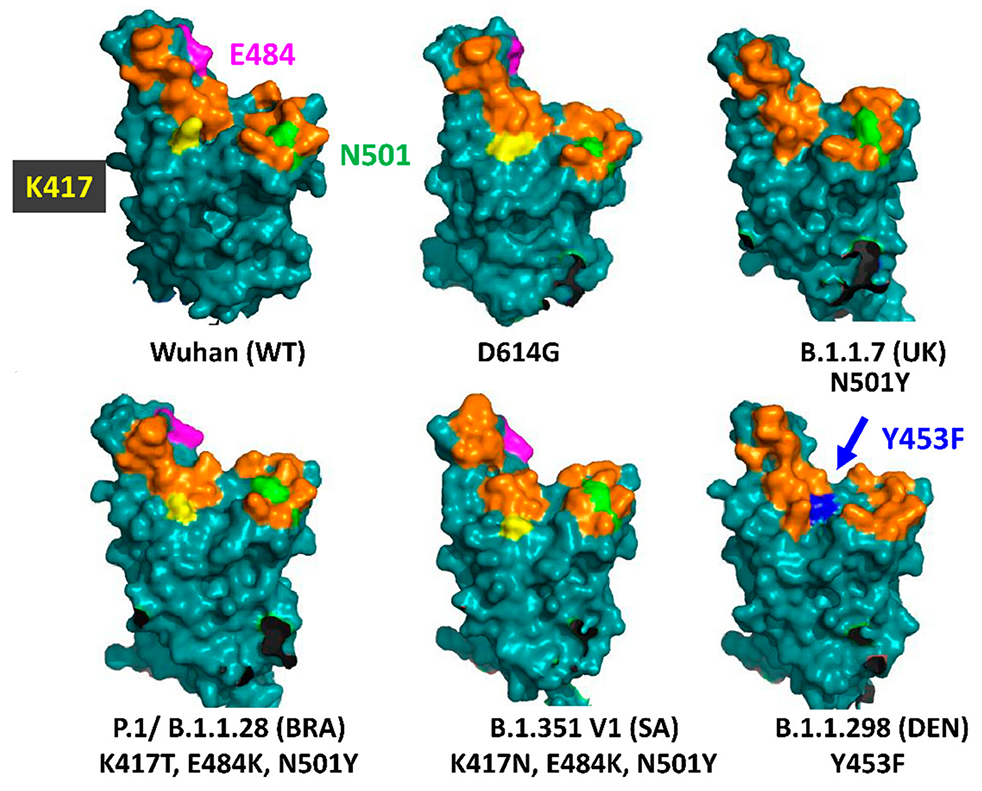
Le variant Alpha comprend 22 mutations par rapport à la lignée ancestrale B.1.1 et 5 délétions, la majorité se trouvant dans la région génomique encodant la protéine S au niveau du domaine de liaison au récepteur (RBD). Plusieurs mutations sont importantes : - La substitution en N501Y3 (N→Y en position 501) est une mutation qui augmente les capacités d'attachement du virus au récepteur ACE2 des cellules humaines et par conséquent elle facilite la propagation du virus. Cette mutation n'aura probablement pas d'effet sur les vaccins actuels. On y reviendra. - Une double délétion aux positions H69-V70. Elle est identique à la mutation qui infecta les visons en novembre 2020 au Danemark. Comme le confirme une étude publiée sur "bioRxiv" le 14 janvier 2021, cette délétion permet au virus de se transmettre plus facilement et donc de plus facilement contaminer les cellules. Comme la délétion Y144-145, elle modifie la forme de la protéine S et peut aider le virus à échapper à certains anticorps monoclonaux et polyclonaux. - La mutation P681H permet aux cellules infectées de fabriquer plus efficacement de nouvelles protéines S. - La mutation A222V sur la protéine S rend les anticorps légèrement moins efficaces. C'est une mutation d'échappement. - La mutation E484K est une mutation sur le domaine RBD de la protéine S. Il s'agit d'une substitution E→K (acide glutamique en lysine) ou E→Q (glutamine) ou E→P (proline). Sa présence sur le variant Alpha fut annoncée officiellement le 1 février 2021 par le PHE. E484K est une mutation d'échappement qui modifie suffisamment la forme du domaine RBD pour permettre au virus d'échapper à certains types d'anticorps monoclonaux et polyclonaux. Les tests montrent qu'elle diminue de 35 à 60 fois la capacité de neutralisation des anticorps anti-RBD chez le sujet contaminé. Cette mutation est identique à celle du variant Gamma découvert au Brésil (voir plus bas). Cette mutation explique en partie la propagation plus rapide de ce variant dans la population que les souches antérieures. Dans la plupart des cas, c'est la présence d'un variant porteur de la mutation d'échappement E4848K qui explique les cas de recontamination. On y reviendra. D'où provient le variant Alpha ? On l'ignore encore. À ce jour, aucun virus étroitement apparenté n'étaye la théorie selon laquelle le variant proviendrait de l'étranger. Sur base des données de décembre, les schémas de mutations observées suggèrent qu'il s'agit d'une évolution adaptative au Royaume-Uni. Des schémas de mutations similaires ont été observés dans l'évolution du SARS-CoV-2 chez des patients infectés chroniquement ayant un système immunitaire affaibli. L'hypothèse actuelle est qu'un tel scénario d'infection chronique chez un seul patient peut avoir joué un rôle dans l'émergence de ce variant.
Soulignons que les délétions particulières identifiées dans la protéine S du variant Alpha sont apparues dans plusieurs autres lignées du virus à une fréquence croissante et sont également observées chez les patients victimes d'infections chroniques où elles peuvent altérer l'antigénicité, c'est-à-dire la reconnaissance par les anticorps immunitaires. Ces délétions peuvent également être associées à d'autres mutations dans la région de liaison de la protéine S du virus, notamment celles observées dans les infections chez les visons d’élevage et à une mutation qui joue un rôle dans la capacité du virus à échapper au système immunitaire humain. Alpha contient également un gène ORF8 tronqué, avec des délétions dans cette région précédemment associées à une diminution de la gravité de la maladie. Au 1 janvier 2021, le variant Alpha était déjà en Europe continentale et s'était propagé dans plus de 30 pays, de la France à la Chine, en passant par l'Islande, les États-Unis et l'Australie notamment. Le 8 janvier 2021, il avait touché 45 pays et continuait à se propager. Le variant dominait en Belgique fin février 2021, une tendance similaire à ce qui fut observé au Royaume-Uni. On observe la même infectiosité accrue pour les variants Beta (B.1.351) et P.1 du fait qu'ils portent plusieurs mutations identiques à celle du variant Alpha. C'est ce variant qui est à l'origine du maintien du taux élevé de contaminations qu'on a observé en Europe depuis début 2021 (cf. les deuxième et troisième vagues épidémiques). Le variant B.1.177 (GB) Le variant B.1.177 est apparu le 14 février 2020 au Royaume-Uni (73%) et s'est ensuite propagé au reste du monde bien qu'il soit surtout représenté en Europe occidentale et en particulier au Royaume-Uni. Il compte actuellement au moins 85 sous-variants généralement associés à une région ou un pays. Le variant B.1.36.17 (GB) Le variant B.1.36.17 est apparu le 16 juillet 2020 au Royaume-Uni (98%) et tend à se propager en Suisse (1%) et ailleurs en Europe ainsi qu'aux Etats-Unis (< 1%). Le variant B.1.36.28 (GB) Le variant B.1.36.28 est apparu le 9 septembre 2020 du Royaume-Uni et reste actuellement isolé à la Grande Bretagne (99%) et Gibraltar (1%). Le variant B.1.221 (NL) Le variant B.1.221 est apparu le 14 février 2020 aux Pays-Bas et s'est rapidement répandu en Europe. Quatre sous-variants propres à l'Allemagne, le Danemark et la Belgique/Luxembourg ont déjà été identifiés. On le signale également en Corée et aux Etats-Unis. Le variant Beta ou B.1.351, également appelé 501.V2 fut découvert à Nelson Mandela Bay, en Afrique du Sud, en octobre 2020. Il est identique au variant britannique Alpha (B.1.1.7). Cependant, les deux mutations sont apparues séparément. En février 2021, le variant Beta s'était propagé dans 32 pays, dont l'est de l'Afrique, la Zambie, le Ghana, aux États-Unis, au Canada, en Europe, en Turquie, en Israël et en Chine. Il tua une personne en Belgique alors qu'elle n'avait pas voyagé.
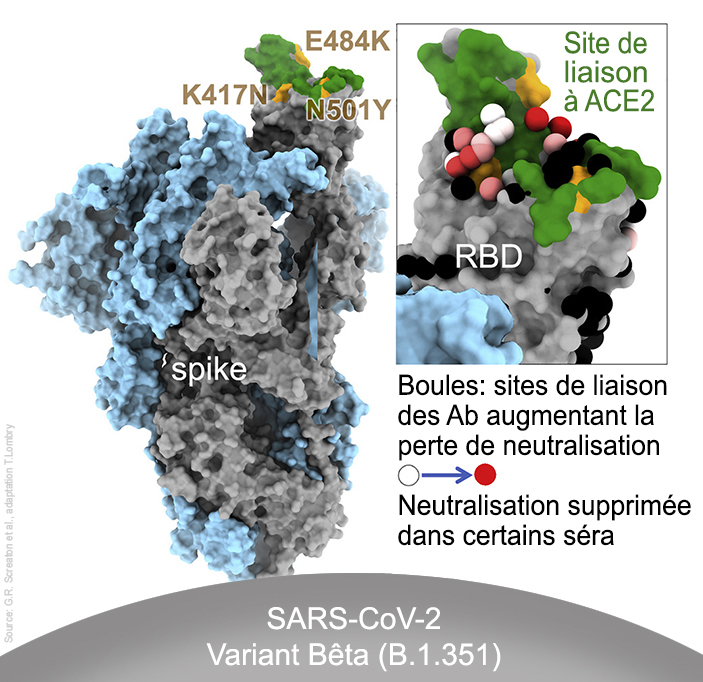
Trois mutations se situent dans le domaine de liaison au récepteur (RBD) de la protéine S : - N501Y aide le virus à s'accrocher plus étroitement aux cellules humaines. Cette mutation apparaît également dans les variants B.1.1.7 et P.1 (cf. P.Dormitzer et al., 2020). - K417N aide également le virus à se lier plus étroitement aux cellules humaines. - E484K peut aider le virus à échapper à certains types d'anticorps monoclonaux et polyclonaux. Ces trois mutations peuvent donc améliorer l'affinité du variant avec le récepteur ACE2 des cellules humaines.
Selon les experts du Global Virus Network américain (GVN), le variant Beta a montré un échappement complet aux anticorps neutralisants dans 48% des échantillons de sérum de convalescence obtenus de patients qui avaient été contaminés par le SARS-CoV-2. Nous verrons page suivante que les vaccins perdent également une partie de leur efficacité face à ce variant. Le variant Beta s'est toutefois relativement peu propagé et finit par être remplacé par des variants plus transmissibles comme Alpha et Delta. Le variant Gamma (P.1, BR) Le variant P.1 ou Gamma, également appelé B.1.1.28.1 (ou encore 20J/501Y.V3) fut découvert par le NIID à Tokyo, au Japon, et est proche du B.1.351. Il fut détecté le 2 janvier 2021 chez quatre personnes de retour au Japon après un séjour dans l'État d'Amazonas (Manaus) situé au nord-ouest du Brésil (Manaus). Il apparut alors que le Brésil subissait une deuxième vague épidémique aussi importante que la première, notamment à Manaus. En octobre 2020, il était le variant dominant (75%) dans les villes d'Amérique latine. Il est également présent aux États-Unis depuis fin janvier 2021, dans plusieurs pays d'Europe et en Turquie. Ce variant porte 14 mutations (et une délétion) dont les principales se situent sur les gènes exprimant la protéine S. Les mutations clés de la protéine S sont similaires à celles du variant B.1.351, bien qu'elles soient apparues indépendamment : - N501Y aide le virus à s'accrocher plus étroitement aux cellules humaines. Cette mutation apparaît également dans les variants B.1.1.7 et B.1.351. - K417T se situe sur le même site que la mutation K417N du variant B.1.351. Elle peut aider le virus à se verrouiller plus étroitement au récepteur cellulaire. - E484K peut aider le virus à échapper à certains types d'anticorps monoclonaux et polyclonaux. Le variant Gamma est présent dans 54 pays mais principalement en Amérique du Nord et au Brésil. Du variant Gamma (P.1) est né le variant B.1.1.248 également nommé VOC-202012/01 qui par la suite a perdu son statut de variant distinct et fut reclassé en B.1.1.28. Selon les chercheurs du NIID japonais, B.1.1.28 présente 12 mutations dans la protéine S dont N501Y et E484K qui modifient légèrement la forme de la protéine S. La mutation à la position E484K est associée à la fuite d'anticorps neutralisants, c'est-à-dire qu'elle permet au virus d'échapper aux cellules mémoires des défenses immunitaires. Du fait de sa capacité à échapper aus défenses immunitaires, il inquiète plus les chercheurs que d'autres variants, d'autant que l'épidémie au Covid-19 a fortement touché la population brésilienne (la barre des 200000 décès fut franchie le 8 janvier 2021) et que cette mutation E484K est également présente au Royaume-Uni. Depuis novembre 2020, le variant Gamma est resté minoritaire mais est présent dans plus de 30 pays dont 15 pays d'Amérique du Sud et en Europe. Le variant L452R (US) Le variant L452R fut découvert au Danemark en mars 2020 et fut ensuite identifié aux Etats-Unis. Selon l'autorité de santé publique de Santa Clara (SSC.gov) en Californie, ce variant est devenu de plus en plus courant dans plusieurs comtés californiens et est responsable de plusieurs foyers épidémiques importants dans l'État. Les résultats du séquençage montrent que la proportion de personnes contaminées par ce variant est passée de 3.8 à 25% entre la mi-novembre et la fin décembre 2020. Selon "The Washington Post", dans l'un des foyers le variant L452R fut transmis à un membre du personnel de l'hôpital de San José portant un costume d'arbre de Noël gonflable qui aurait pu infecter au moins 90 personnes. Selon Erica Pan, épidémiologiste au Département de la santé publique de Californie, "Il est trop tôt pour savoir si cette variante se répandra plus rapidement que d'autres". En février 2021, cette mutation était présente chez ~45% des échantillons récoltés en Californie. Le variant Iota (B.1.526, US) Le variant B.1.526 ou Iota fut identifié à New York le 21 avril 2020. Il possède notamment les mutations de pointe T95I et D253G parmi d'autres. Une plus petite fraction des porteurs de ce variant ont S477N au lieu de E484K. La mutation Spike E484K est présente dans environ la moitié de cette lignée ainsi que E484K et S477N qui le rapproche du variant Lambda alias C.37 (voir plus bas). Le variant Iota est présent en Amérique du Nord et du Sud ainsi qu'en Europe. Le variant B.1.429 (US) Le variant B.1.429 est apparu en Californie, dans la banlieue de Los Angeles, en juillet 2020 et est responsable de la vague épidémique qui survint dans la mégapole. Depuis, il s'est répandu dans 26 pays, y compris en Europe où on a identifé quelques cas, notamment en Angleterre, en France et en Scandinavie. Ce variant porte 10 mutations dont certaines sur les acides aminés S13I, W152C, L452R et D614G. Les mutations L452R et D614G réduisent la capacité de neutralisation des anticorps et donc augmentent le pouvoir infectieux du virus. Le variant B.1.427 (US) Le variant B.1.427 fut identifié fin septembre 2020 en Californie, il est présent dans toute l'Amérique du Nord et dans 14 pays autour dans le monde, dont l'Angleterre et l'Italie. Ce variant porte 8 mutations dont certaines sur les acides aminés L452R et D614G. Les mutations L452R et D614G réduisent la capacité de neutralisation des anticorps et donc augmentent le pouvoir infectieux du virus. Le variant COH.20G/501Y (US) Le variant COH.20G/501Y fut découvert aux États-Unis en décembre 2020. Selon le College of Medicine, il porte une mutation identique au variant britannique B.1.1.7, mais elle est probablement apparue dans un variant qui était déjà présent aux États-Unis. Les chercheurs signalent également l'évolution d'un autre variant américain qui développa trois autres mutations géniques qui n'avaient jusqu'ici jamais été observées ensemble dans le SARS-CoV-2. La découverte de ce variant dit de Columbus fit l'objet d'une article publié sur "bioRxiv" (non validé) par l'équipe de Dan Jones le 15 janvier 2021. On ignore encore sa prévalance dans la population. En revanche, il est devenu le virus dominant à Columbus, Ohio, pendant une période de trois semaines, entre fin décembre 2020 et janvier 2021. Selon Jones, "ces trois mutations [dans le variant de Columbus] représentent une évolution significative. Nous savons que ce changement n'est pas venu des branches britanniques ou sud-africaines du virus. [...] Les changements observés au cours des deux derniers mois ont été plus importants que dans les premiers mois de la pandémie." Les chercheurs suggèrent que la même mutation aurait pu se produire indépendamment dans plusieurs régions du monde au cours des derniers mois (c'est-à-dire depuis novembre 2020). Le variant B.1.617 (IN) Le variant B.1.617 fut découvert en octobre 2020 près de Nagpur, en Inde. En avril 2021, il représentait en moyenne ~11% des contaminations dans le pays mais sa prévalence atteint 55% à Nagpur et Mumbai. Le variant était également présent au Royaume-Uni, en Irlande, en Allemagne, et en Belgique, mais avec une très faible occurrence. Ce variant présente 15 mutations dont deux connues, L452R identique à celle du variant californien qui le rend plus résistant aux anticorps et E484Q très proche de la mutation observée sur les variants brésilien et sud-africain qui le rende plus contagieux. C'est la première fois qu'on identifie ces deux mutations sur le même variant. Apparement ce variant est moins infectieux que le variant B.1.177 britannique. Du variant B.1.617 sont nées trois lignées : B.1.617.1 (Kappa) et B.1.617.2 (Delta) qui donna le sous-variant B.1.617.3. Le variant Delta (B.1.617.2, IN) Le variant Delta (B.1.617.2 ou G/452R.V3) fut détecté au Maharashtra, au centre de l'Inde, fin 2020. Il présente 17 mutations dont 2 délétions. Les mutations les plus importantes sur la protéine S sont L19R, une délétion à la position 157/158, L452R et T478K dans le domaine N-terminal, et D950N dans le domaine de liaison au récepteur (RBD). Il ne présente pas la mutation E484Q (cf. F.M. Giorgi et al., 2021; ProteoGenix). Notons que c'est la mutation L452K qui est recherchée dans les tests de dépistage pour confirmer la présence du variant Delta dans un échantillon.
L'OMS a prévenu les autorités nationales que le variant Delta serait environ 40% puis finalement 60% plus transmissible que le variant Alpha (B.1.1.7) qui est lui-même 60% (entre 35 à 90%) plus contagieux que la souche originale. De nouvelles données indiquent qu'il est 3 fois plus transmissible que la souche de Wuhan. Selon l'autorité australienne de la santé (cf. Reuters, 5 juin 2021) le variant Delta est "hautement infectieux" et est "50% plus transmissible que le variant Alpha". Il présente une charge 10 fois plus élevée que le variant Alpha. Une étude publiée le 7 juillet 2021 montre que la charge du variant Delta est ~1000 fois plus élevée et qu'il se propage 2.25 fois plus vite que le variant Alpha. Selon le PHE britannique, le variant Delta augmente de 2.61 fois le risque d'hospitalisation dans les 14 jours par rapport au variant Alpha. Enfin, selon une étude canadienne publiée le 25 octobre 2021, le variant Delta est 133% plus mortel que le virus de Wuhan.
En résumé, le variant Delta est aussi contagieux que la varicelle dont le taux de reproduction de base Ro varie entre 10 et 12. Le simplement croisement avec une personne contaminée durant 5 secondes transmet le variant Delta. Par conséquent les porteurs du variant Delta sont considérés comme des super-contaminateurs. Le variant Delta peut également échapper à certains vaccins mais toutes les études ont montré que les vaccins à deux doses nous protègent également contre ce variant. On y reviendra. Ce variant Delta serait responsable de la deuxième vague épidémique survenue en Inde en février 2021. Il est présent en Europe et est devenu le variant dominant au Royaume-Uni (plus de 90% des cas fin juin 2021) mais resta quelque temps minoritaire en Belgique et en France notamment tout en présentant plus de 30% des contaminations. Depuis juillet 2021, il est dominant avec par exemple près de 95% des contaminations en Belgique. Il est également dominant aux Etat-Unis depuis juillet 2021 avec 51% des nouvelles contaminations. Début juillet 2021, le variant Delta était présent dans 85 pays. Sa propagation rapide reste préoccupante et est à l'origine de la 4e vague épidémique dans certains pays européens notamment, principalement véhiculée par les jeunes et les personnes revenant de vacances de pays en zone rouge. Le variant Delta pose un problème de santé publique car étant plus transmissible et plus contagieux, il augmente le Ro du virus jusque 6 et parfois jusque 10 et par conséquent la couverture vaccinale doit concerner plus de personnes pour atteindre l'immunité collective. Jusque fin 2020, en Europe pour un Ro ~3 on estimait que ~70% de la population devait être vaccinée pour être globalement protégée. Avec ce variant, Ro > 6 et on devrait vacciner plus de 83% de la population. Depuis l'automne 2021, plusieurs ministres de la santé et des chefs de gouvernements européens envisagent une couverture vaccinale de 90 voire 100% de la population pour enrayer la propagation de la pandémie. En septembre 2021, nous étions loin en dessous de cet objectif (cf. les campagnes de vaccination). Selon l'OMS, "Il est devenu évident que davantage de risques pour le public sont associés au B.1.617.2" (via ONU). Le variant Delta plus Le 5 avril 2021, une mutation du variant Delta nommée "Delta plus" (B.1.716.2.1) fut identifiée au Maharashtra, au centre de l'Inde. Fin juin 2021, il s'était déjà propagé jusqu'en Europe. Comme le variant Beta, le nouveau variant Delta plus porte la mutation K417N sur la protéine S où une lysine a été remplacée par une asparagine qui lui permet d'échapper aux défenses immunitaires. Selon Ravindra Gupta, immunologiste à l'Université de Cambridge, la mutation K417N ne devrait pas modifier fondamentalement le variant Delta plus, ni le rendre plus dominant. Selon l'OMS, Delta plus n'est pas différencié de Delta. Le variant Lambda (C.37, PE) Le variant C.37 alias B.1.1.1.37 ou Lambda fut identifié au Pérou le 8 novembre 2020 où il fut rapidement dominant et s'est ensuite rapidement propagé en Amérique latine, notamment au Brésil. En juillet 2021, il était présent en Amérique du Nord, en Europe (Allemagne, Espagne, France, Belgique, Royaume-Uni, etc) et en Australie. Selon Jeff Barrett, directeur de la Covid-19 Genomics Initiative au Wellcome Sanger Institute de Londres, ce variant "présente un ensemble inhabituel de mutations" comparé aux autres variants. Le variant Lambda présente les 8 mutations de la lignée C.37 (gène ORF1a : 3675-3677 ; gène Spike : Δ246-252, G75V, T76I, L452Q, F490S, D614G et T859N) en plus de 19 autres mutations. Classé parmi les VOI, il a été associé à des taux élevés de transmissibilité (cf. P.L. Wink et al., 2021). Dans son rapport de la mi-juin 2021, l'OMS signale que le variant "Lambda a été associée à des taux substantiels de transmission communautaire dans plusieurs pays, avec une prévalence croissante au fil du temps parallèlement à une incidence accrue de Covid-19" et que davantage d'études seraient menées sur ce variant. Selon le virologue Emmanuel André de l'UCL, le variant Lambda semble plus faible que les variants Gamma et Delta mais les avis sont encore partagés. Deux mutations, T76I et L452Q, contribuent à rendre le variant Lambda très infectieux. Selon une étude de l'Université de Santiago du Chili publiée sur "medRxiv" le 1 juillet 2021, le variant Lambda pourrait être plus contagieux que d'autres variants : "on a observé une augmentation de l'infectivité médiée par la protéine de pointe [du variant] Lambda qui était encore plus élevée que celle du D614G (lignée B) ou des variants Alpha et Gamma. Par rapport au type sauvage (lignée A), la neutralisation a diminué de 3.05 fois pour le variant Lambda alors qu'elle était de 2.33 fois pour le variant Gamma et de 2.03 fois pour le variant Alpha". Selon un article publié par des chercheurs japonais sur "bioRxiv" le 28 juillet 2021, les trois mutations RSYLTPGD246-253N, 260 L452Q et F490S situées sur la protéine S peuvent conférer au varian Lambda une résistance face à l'immunité induite par les vaccins, mais des recherches supplémentaires sont nécessaires. Le variant Eta (B.1.525, WAN) Le variant B.1.525 ou Eta fut détecté au Nigéria le 11 décembre 2020. Il est présent dans 61 pays, y compris en Europe et en Amérique du Nord. Il contient deux délétions (H69-V70 et Y144) sur la protéine S commune avec le variant Alpha (B.1.1.7) et la mutation E484K commune aux variants Beta (B.1.351) et Gamma (P.1) qui permet au virus d'échapper aux cellules mémoires des défenses immunitaires. Il comprend également des substitutions d'acides aminés Q52R, Q677H et F888L73. Des substitutions répétées d'acides aminés en position 677 et l'émergence indépendante de Q677H dans plusieurs variants aux États-Unis sont des indices forts de l'adaptation du virus (cf. D.L. Robertson et al., 2021). Le variant Mu (B.1.621, CO) Le variant B.1.621 alias 21H ou Mu fut identifié en Colombie en janvier 2021. Il était déjà présent au Mexique, au Chili et en Espagne. Selon le CDC américain, ce variant représente 10% des échantillons positifs dans le sud de la Floride et 2% des échantillons de l'ensemble des Etats-Unis. A ce jour, il est présent dans 39 pays. Vingt souches ont été isolées dont B.1.621.1 qui est présent depuis avril 2021 aux Etats-Unis, en République Dominicaine, en Espagne, en Autriche et en Suisse. Le variant Mu présente les mutations D614G, P681H et R346K sur la protéine S ainsi que E484K, N501Y et K417N dans le domaine RBD de la protéine S. Le variant Mu partage les mutations E484K et K417N avec le variant Beta. Il peut donc s'accrocher plus étroitement aux cellules humaines et échapper aux défenses immunitaires. Malgré ces avantages, le variant Mu semble moins performant que le variant Delta et a peu de chance en théorie de le remplacer. Les premiers résultats in vitro montrent que les anticorps induits par la vaccination contre le Covid-19 ou suite à une infection antérieure ont moins de chances de neutraliser le variant Mu. Cependant, ces résultats doivent encore être confirmés par d'autres études. Le degré exact de transmission du variant Mu n'a pas encore été déterminé.Selon une étude britannique basée sur seulement quelques échantillons, ce variant ne serait pas plus dangereux ni plus contagieux que les variants apparus depuis fin 2020. Mais nous manquons de données pour l'affirmer. Le 21 juillet 2021, le PHE nota que le variant ne semble pas se propager particulièrement rapidement et il est "peu probable" qu'il soit plus transmissible que le variant Delta. Par conséquent, selon le PHE, "il n'y a aucune indication que [Mu] surpasse Delta" pour le moment. Mais la capacité du variant à échapper aux défenses immunitaires induites par la vaccination "pourrait contribuer à de futurs changements de croissance". Début août 2021, le variant Mu provoqua sept décès en Belgique (cf. RTBF). Il est présent en Espagne, en Italie, en France et en Suisse. En septembre 2021, il représentait 39% des cas de contamination en Colombie et 13% des cas en Equateur. A l'échelle mondiale, il ne représente que 0.1% des séquences analysées. Le variant C1.2 (ZA) Le variant C.1.2 fut identifié en mai 2021 dans les provinces de Gauteng et Mpumalanga, en Afrique du Sud. Il évolua à partir de la souche C.1 qui domina durant la première vague épidémique en Afrique du Sud (cf. C.Scheepers et al., 2021). Depuis, il s'est propagé dans sept autres pays d'Afrique, en Europe, en Asie et en Océanie. Lors d'un briefing qui s'est tenu au siège de l'ONU à Genève le 31 août 2021, Margaret Harris, porte-parole de l'OMS, déclara qu'il faut encore prouver que ce variant présente "une transmissibilité, une virulence ou un changement clinique accrus de la maladie, et une efficacité réduite des mesures de santé publique et sociales" (cf. Reuters). Or l'étude précitée de l'équipe de Scheepers publiée le 26 août 2021 (non validée) indique que le variant C.1.2 contient plusieurs substitutions (R190S, D215G, E484K, N501Y, H655Y et T859N) et délétions (Y144del, L242-A243del) dans la protéine S. Selon les chercheurs, "Ces mutations sont associées à une transmissibilité accrue et une sensibilité de neutralisation réduite. L'accumulation de mutations supplémentaires (C136F, Y449H et N679K) est plus préoccupante, car elles sont susceptibles d'avoir un impact sur la sensibilité à la neutralisation par les anticorps ou le clivage de la furine et donc la capacité de réplication". Le taux de mutation du variant C.1.2 est deux fois plus élevé que celui des autres variants avec 41.8 mutations par an. C'est actuellement le variant le plus éloigné de la souche originale du SARS-CoV-2 identifiée à Wuhan au début de la pandémie. Ceci dit, il existe beaucoup d'autres variants mais leur proportion est généralement très faible. Ils se concentrent dans certaines villes voire même dans certaines populations, d'où la nécessité de dépister tout le monde et périodiquement. Le variant Omicron (B.1.1.529, BW) Le variant B.1.1.529 ou Omicron fut identifié dans des échantillons collectés le 11 novembre 2021 au Botswana et le 14 novembre 2021 dans la région de Johannesbourg, en Afrique du Sud. Soulignons que le clade 21M d'Omicron avait plus de 4800 descendants au 1 janvier 2022. Si on remonte son arbre phylogénétique jusqu'à son ancêtre B.1.1, on constate qu'il avait déjà subi des mutations en mars 2020 (clade 20B) mais qui n'avaient pas touché ses acides aminés. Cela signifie qu'il ne fut pas détecté à l'époque. D'où l'intérêt de développer des modèles prédictifs des futurs variants (cf. le modèle DCA décrit précédemment) afin que les microbiologistes, les épidémiologistes et les sociétés pharmaceutiques puissent anticiper l'émergence d'un nouveau variant et d'une éventuelle vague épidémique. Omicron s'est rapidement propagé dans les pays limitrophes (Lesotho, Zimbabwé, etc) puis en Europe via les transports aériens. Il fut d'abord détecté en Belgique et au Royaume-Uni chez des passagers revenant d'Afrique du Sud qui furent aussitôt mis en quarantaine. On demanda aux voyageurs rentrant d'Afrique du Sud d'effectuer un test PCR au deuxième jour de leur retour. En 5 jours, le variant Omicron était présent au Pays-Bas, en Allemagne, en Suisse, en Italie, en France, au Danemark, à Hong Kong, en Australie et au Canada. En moins d'une semaine, Omicron s'était propagé à travers le monde. Mi-décembre 2021, il était présent dans 65 pays.
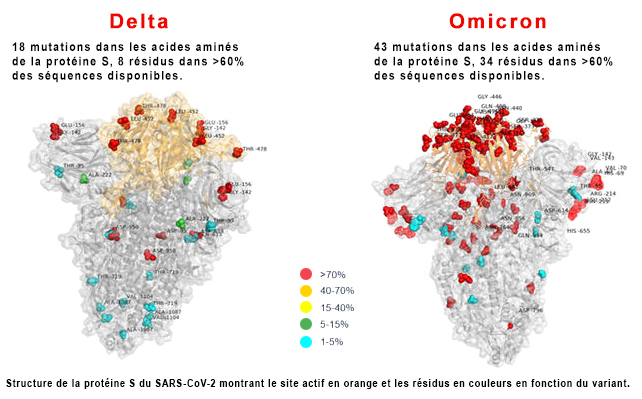
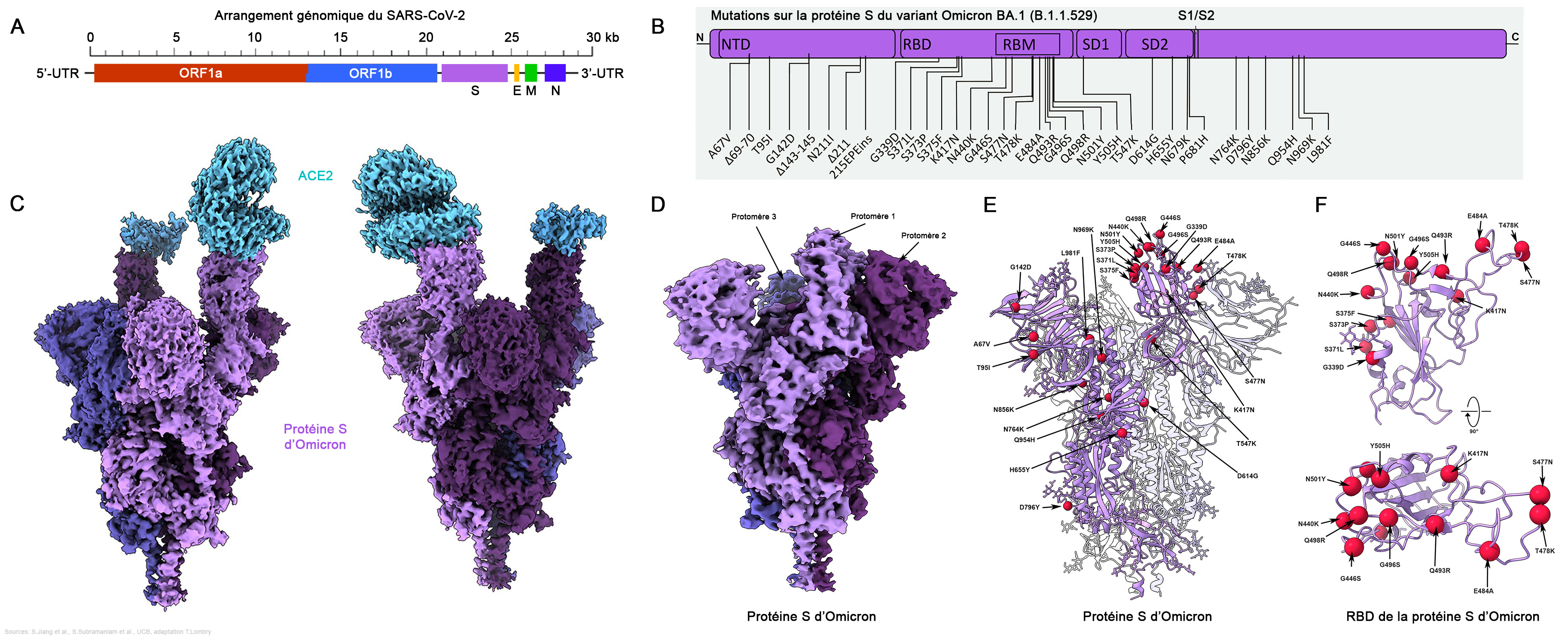
Selon l'ECDC, du fait de la présence du variant Delta, "le niveau de risque global pour l'Union Européenne associé au variant Omicron du SARS-CoV-2 est évalué comme élevé à très élevé". Ce risque élevé se maintiendra probablement jusqu'en février ou mars 2022. Par précaution, pour éviter de nouvelles contaminations, plusieurs pays européens ainsi que les Etats-Unis et Israël parmi d'autres ont annulé tous les vols provenant d'Afrique du Sud. Plusieurs pays ont également fermé leurs frontières dont les Etats-Unis, Israël et le Japon, ce qu'ont regretté Cyril Ramaphos, le président de l'Afrique du Sud, et l'OMS (cf. Reuters). Seuls les ressortissants nationaux peuvent rentrer dans leur pays. Selon des estimations établies en janvier 2022 par l'Institute for Health Metrics and Evaluation (IHME), plus de 60% de la population mondiale a été exposée à Omicron et plus de 65% de la population mondiale a reçu au moins une dose du vaccin. On estime que cette immunité acquise devrait ralentir le développement de nouveaux variants ou sous-variants. Selon le rapport de l'ECDC publié le 26 novembre 2021, le variant Omicron appartient à la lignée Pango B.1.1.529, clade Nextstrain 21K, et est caractérisé par 37 mutations sur la protéine S dont 30 changements d'acides aminés, trois délétions et une insertion par rapport au virus d'origine de Wuhan (A67V, 69-70, T95I, G142D, 143-145, Δ211, L212I, ins214EPE, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493K, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, L981F). Parmi ces changements, 15 sont situés dans le domaine de liaison au récepteur (RBD) (résidus 319-541). Le variant comporte également un certain nombre de changements et des délétions dans d'autres régions génomiques (NSP3 – K38R, V1069I, Δ1265, L1266I, A1892T ; NSP4 – T492I ; NSP5 – P132H ; NSP6 – Δ105-107, A189V ; NSP12 – P323L ; NSP14 - I42V ; E-T9I ; M-D3G, Q19E, A63T ; N – P13L, Δ31- 33, R203K, G204R). Au total, depuis sa divergence de la lignée B.1.1, fin décembre 2021 Omicron a subi 45 mutations.
Le variant Omicron est le variant le plus divergent qui ait été détecté à ce jour au cours de la pandémie au Covid-19, ce qui soulève de sérieuses inquiétudes sur l'efficacité des vaccins et le risque potentiellement accru de réinfection. Plusieurs des changements génétiques dans la séquence codant la protéine S ont été décrits auparavant et sont associés à une transmissibilité accrue, à une évasion immunitaire ou à d'autres propriétés. Dans un article publié dans la revue "Science" le 20 janvier 2022, l'équipe de Sriram Subramaniam de l'Université de Colombie Britannique réalisa une analyse structurelle de ces mutations. Les chercheurs ont montré que plusieurs mutations (Q493K, G496S et Q498R) créent de nouveaux ponts salins et des liaisons hydrogène entre la protéine S et le récepteur ACE2 cellulaire. Les chercheurs ont conclu que ces nouvelles liaisons semblent augmenter l'affinité de liaison - la force avec laquelle le virus se fixe aux cellules humaines - tandis que d'autres mutations (K417N) diminuent la force de cette liaison. Selon Subramaniam, "Dans l'ensemble, les résultats montrent qu'Omicron a une plus grande affinité de liaison que le virus d'origine, avec des niveaux plus comparables à ce que nous voyons avec le variant Delta. Il est remarquable que le variant Omicron ait évolué pour conserver sa capacité à se lier aux cellules humaines malgré des mutations aussi importantes". En examinant ces mutations, un certain nombre rendent vraisemblablement le variant plus infectieux. D'autres mutations sont encore plus inquiétantes, car elles interférent avec la capacité du système immunitaire à reconnaître le virus. Subramaniam et ses collègues ont montré qu'Omicron dispose d'une évasion mesurable pour six anticorps monoclonaux testés, avec une évasion complète pour cinq d'entre eux. Le variant affiche également une évasion accrue des anticorps collectés auprès d'individus vaccinés et de patients Covid non vaccinés. On reviendra sur les raisons pour lesquelles un virus et notamment Omicron peut échapper aux défenses du système immunitaire. Selon Subramaniam, "Omicron est moins évasif face à l'immunité induite par les vaccins comparée à l'immunité naturelle contre la contamination chez les patients non vaccinés. Cela suggère que la vaccination reste notre meilleure défense". On y reviendra (cf. page 4). Un variant synthétique précédemment décrit avec 20 mutations dans la protéine S a été associé à une évasion quasi complète des sérums de convalescence et des vaccinés. Comme Omicron porte encore plus de mutations sur la protéine S que les autres variants, cela a effet très important sur la neutralisation. On reviendra page suivante sur l'efficacité des vaccins. Sur la base de l'augmentation observée de l'affinité de liaison et de l'évasion des anticorps, les chercheurs affirment que les mutations de la protéine S sont probablement des facteurs contribuant à l'augmentation de la transmissibilité du variant Omicron. En effet, dès les premières analyses on a constaté que ce variant est 3 fois plus transmissible que le variant Delta qui est lui-même 3 fois plus transmissible que la souche originale de Wuhan. Selon l'OMS, Omicron est le variant le plus transmissible du SARS-CoV-2.
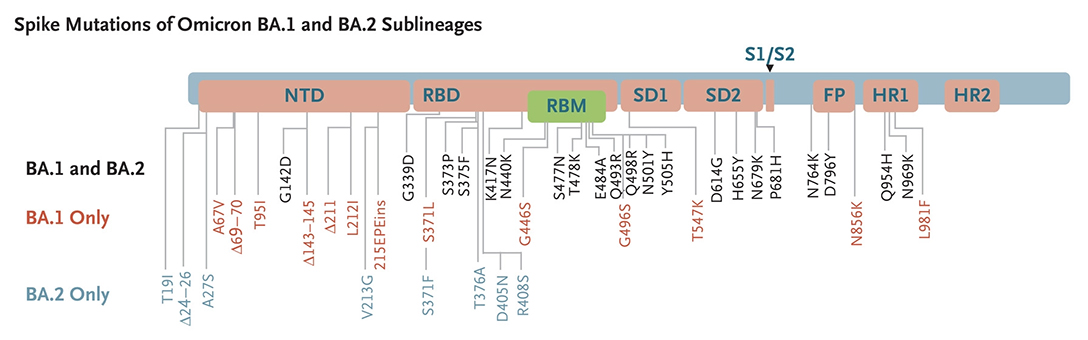
Concernant la gravité d'une éventuelle infection, les informations préliminaires provenant d'Afrique du Sud indiquaient qu'aucun symptôme inhabituel n'a été associé à Omicron, et comme d'autres variants, certains individus sont asymptomatiques. Omicron touche peu les jeunes qui n'éprouvent que des symptômes bénins. Dans une étude publiée dans la revue "Nature Research Briefings" le 23 décembre 2021, l'équipe de Davide Corti de Humabs Biomed, une filiale de Vir technologies, a développé un pseudovirus inactif du SARS-CoV-2 et découvert que la protéine S du variant Omicron se lie 2.4 fois plus solidement au récepteur ACE2 que la souche originale de Wuhan et que les porteurs contaminés par les anciens variants sont moins bien immunisés contre le variant Omicron. Dans un rapport publié par l'agence de Sécurité Sanitaire britannique (UKHSA) le 29 décembre 2021, les chercheurs constataient que le risque d'admission à l'hôpital en soins intensifs avec Omicron représentait environ un tiers du risque constaté avec le variant Delta. Cependant les auteurs soulignent l'augmentation très importante du nombre de contaminés que tous les pays doivent anticiper en prenant les mesures préventives adéquates. On y reviendra à propos de l'incidence des cas de contamination. Les sous-variants d'Omicron BA.1, BA.2, etc. En décembre 2021, les microbiologistes ont découvert un sous-variant d'Omicron nommé BA.2 qui contient quelques mutations supplémentaires. On découvrit rapidement qu'il existait deux sous-variants, BA.1 et BA.2 (cf. D.Yamasabo et al., 2022). Plus récemment on découvrit le sous-variant BA.3 puis BA.1.1 qui présente une substitution du gène codant pour la protéine S.
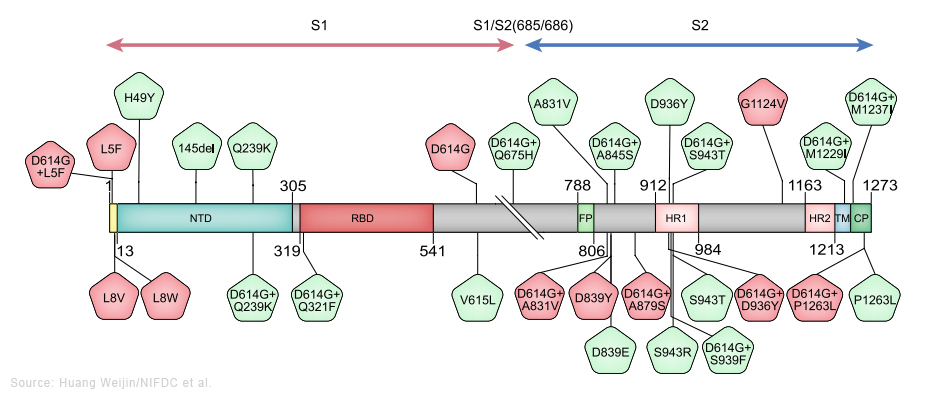
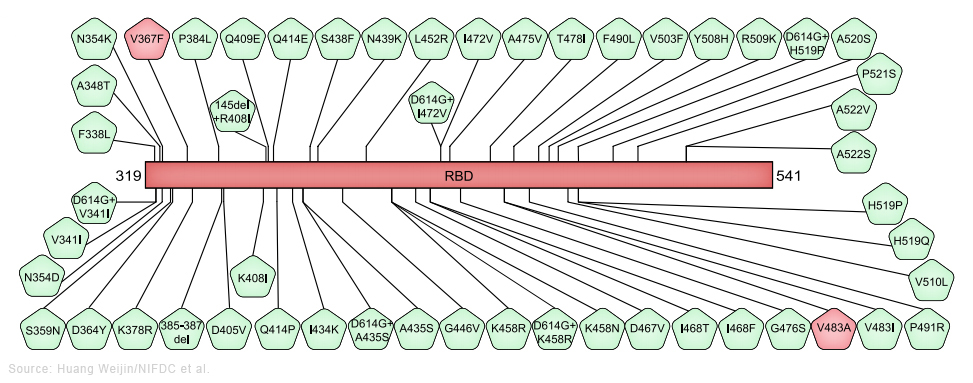
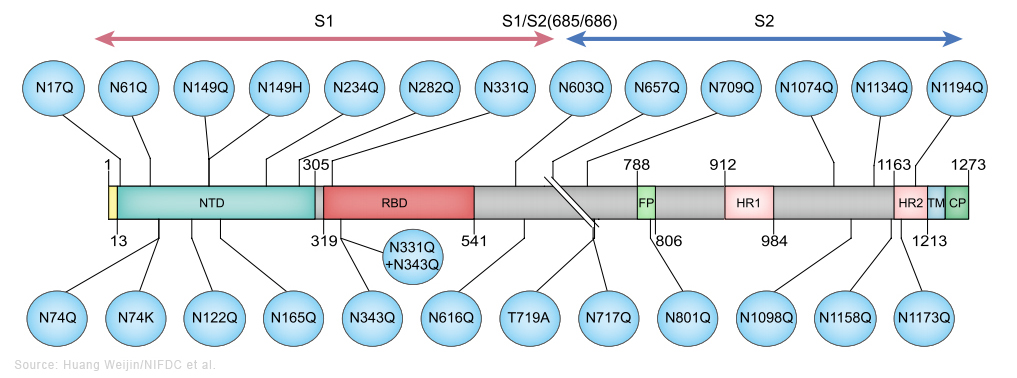
Le sous-variant BA.2 est considéré comme un variant d'Omicron "furtif" car selon les chercheurs il pourrait être plus difficile à traquer que le variant original. Contrairement à Omicron, BA.1 porte une délétion dans le gène qui code pour la protéine S. Cette mutation spécifique fait que les tests PCR affichent un message d'erreur : "S gene target failure" (Échec de la cible du gène S) lorsqu'ils détectent l'Omicron d'origine. Ce message d'erreur a rendu le variant BA.1 facile à suivre lors de sa première apparition. Notons que ce message d'erreur n'a pas affecté la capacité des tests PCR à détecter une personne contaminée par le SARS-CoV-2 car les tests recherchent plusieurs gènes du coronavirus. Le sous-variant BA.2 ne présente pas cette mutation; il ne génère donc pas le même message d'erreur. Cela signife, au moins sur les tests PCR, que cet Omicron furtif ressemble au variant Delta parmi d'autres ne possédant pas cette mutation sur la protéine S. Aussi, pour identifier correctement le sous-variant BA.2 parmi les différents variants en circulation, les chercheurs devaient originellement effectuer une analyse génomique complète. Depuis mars 2022, les sous-variants d'Omicron représentent la grande majorité des nouveaux cas dans le monde. Avec peu ou pas de cas liés au variant Delta ou d'autres variants, même furtif, le BA.2 devrait être plus facile à détecter. Jusqu'à présent l'impact des sous-variants d'Omicron sur la population et sur le système de santé reste modéré et inférieur aux vagues épidémiques précédentes - la vaccination y a contribué -, sauf en Chine. Selon Reuters qui rapporte les propos du Dr John Nkengasong, directeur du CDC Africa, des études montrent que BA.2 est environ 30% plus transmissible que BA.1, bien que selon l'OMS cette différence de contagiosité ne soit pas aussi importante qu'entre Delta et Omicron. Selon Nkengasong ainsi que les résultats d'une étude danoise publiée le 20 janvier 2022, bien que BA.2 semble être plus contagieux que BA.1, il ne provoque pas de formes plus grave de la maladie ni plus d'hospitalisations que le sous-variant BA.1. Depuis, d'autres sous-variants d'Omicron sont apparus : BA.2.12.1, BA.3, BA.4 et BA.5. Ils diffèrent tous les uns des autres par des mutations différentes dans la protéine S. Les sous-variants BA.4 et BA.5 sont responsables de la nouvelle épidémique apparue en mai-juin 2022, notamment en Europe. Nous n'avons aucune preuve suggérant qu'ils seraient plus inquiétants que l'Omicron original, autre qu'une légère augmentation potentielle de la transmissibilité (cf. T. de Oliveira et al., 2022). Selon l'institut Pasteur, BA.5 serait 10% plus contagieux que BA.2, raison pour laquelle il devient dominant. Il est à l'origine de la 8e vague épidémique de Covid-19 en Europe. On reviendra page suivante sur l'efficacité des vaccins et la neutralisation des sous-variants BA.1 et BA.2. Les sous-variants BQ.1 et BQ.1.1. d'Omicron BQ.1 est un sous-variant de BA.5 qui porte des mutations sur la protéine S dans certains sites antigéniques clés, notamment K444T et N460K. En plus de ces mutations, le sous-variant BQ.1.1 porte une mutation de pointe supplémentaire dans un site antigénique clé nommé R346T. Selon l'OMS, entre le 3 et le 9 octobre 2022, d'après les séquences soumises au GISAID, BQ.1 présentait une prévalence de 6% et était présent dans 65 pays. Bien qu'il n'y ait pas de données sur la gravité ou l'évasion immunitaire des études chez l'être humain, BQ.1 montre un avantage de croissance significatif par rapport aux autres sous-variants d'Omicron en circulation dans de nombreux contextes, y compris en Europe et aux États-Unis, et justifie donc une surveillance étroite. Le risque de réinfection plus élevé est une possibilité qui nécessite une enquête plus approfondie. En novembre 2022 aucune donnée épidémiologique ne suggérait une augmentation de la gravité de la maladie. L'impact des modifications immunologiques observées sur la fuite vaccinale reste à établir. Sur la base des connaissances actuellement disponibles, la protection par les vaccins (à la fois l'index et les vaccins bivalents récemment introduits) contre l'infection peut être réduite, mais aucun impact majeur sur la protection contre les maladies graves n'est attendu. Selon une étude sur les hamsters publiée le 5 décembre 2022 sur "bioRxiv" (non validée), les auteurs confirment les données antérieures concernant le sous-variant BQ.1.1. Ce sous-variant qui est à l'origine la 9e vague épidémique de Covid-19 en Europe présente un très haut niveau de résistance aux anticorps neutralisants, ce qui facilite sa propagation. Selon les auteurs, de tous les virus étudiés depuis le début de la pandémie, celui-ci montre "la meilleure forme physique". Ceci dit, il est moins pathogène qu'Omicron BA.5 (cf. le tweet d'Eric Topol). Le sous-variant XBB* d'Omicron XBB* est un sous-variant recombinant (voir plus bas) des sous-variants BA.2.10.1 et BA.2.75 d'Omicron. Selon l'OMS, entre le 3 et le 9 octobre 2022, d'après les séquences soumises au GISAID, XBB* présentait une prévalence globale de 1.3% et était présent dans 35 pays. La prévalence de XBB* varie d'une région à l'autre, mais elle n'a pas encore été systématiquement associé à une augmentation des nouvelles infections. Bien que d'autres études soient nécessaires, les données actuelles ne suggèrent pas qu'il existe des différences substantielles dans la gravité de la maladie pour les infections par XBB*. Il existe cependant des preuves précoces indiquant un risque de réinfection plus élevé, par rapport aux autres sous-variants d'Omicron en circulation. Les cas de réinfection ont été principalement limités aux personnes ayant subi une infection initiale dans la période pré-Omicron. À l'heure actuelle (novembre 2022), il n'existe aucune donnée sur son éventuel échappement immunitaire comme c'est le cas pour d'autres sous-variants d'Omicron. L'impact potentiel des sous-variants BQ.1* et XBB* est fortement influencé par le paysage immunitaire régional. Selon un communiqué de l'OMS publié le 27 octobre 2022, une "soupe" des sous-variants BQ.1.1 et XBB d'Omicron pourraient être à l'origine de la prochaine vague de Covid-19. On reviendra sur les vagues épidémiques liées aux sous-variants d'Omicron, notamment aux Etat-Unis, en Europe et en Chine. Le variant Deltacron (CY) Le 8 janvier 2022, Bloomberg annonça que des chercheurs chypriotes avaient découvert à Chypre en décembre 2021 un nouveau variant recombinant (voir plus bas) du SARS-CoV-2. Leondios Kostrikis, professeur de biologie à l'Université de Chypre et responsable du Laboratoire de Biotechnologie et de Virologie moléculaire le nomma "Deltacron". Selon Kostrikis, il présente le génome du variant Delta mais contient des signatures génétiques similaires à Omicron. Kostrikis et ses collèges prétendent qu'ils ont identifié 25 cas. Mais ils ignorent l'effet de ce variant, s'il conduit à plus de cas de contamination, s'il est plus contagieux ou phatologique. Ils ne savent pas non plus si le nouveau variant supplantera les autres variants tels que Delta et Omicron. Mais cette découverte fut critiquée par d'autres chercheurs qui soupçonnent une erreur de manipulation en laboratoire plutôt qu'un véritable nouveau variant. D'autres chercheurs ont examiné les détails génétiques du prétendu variant Deltacron mis en ligne dans la base de données GISAID (Nextstrain) et ont déclaré qu'il ne ressemble pas à un variant recombinant et que par conséquent il s'agit probablement d'une contamination survenue en laboratoire. Kostrikis a rejeté les accusations de mauvaises manipulations dans son laboratoire. Mais Tom Peacock, virologue au Département des maladies infectieuses de l'Imperial College de Londres, a examiné ce génome et déclara le 21 décembre 2022 sur Twitter qu'il pense qu'il s'agit d'une contamination ou d'une coinfection. Il doute de la prévalence de tout variant recombinant d'Omicron du fait que ce dernier circule depuis peu de temps. Dans un second tweet publié le 8 janvier 2022, Peacock expliqua qu'une erreur de manipulation est très certainement à l'origine du variant recombinant : "Ce n'est pas vraiment lié à la qualité du laboratoire ou à quelque chose de similaire. Cela arrive littéralement à chaque laboratoire de séquençage occasionnellement ! Cela est particulièrement vrai avec des écouvillons à faibles niveaux de virus". De plus, si on examine les arbres phylogénétiques dans Nextstrain, toutes les séquences des virus ont un ancêtre commun. Or pour Deltacron, ce n'est pas le cas. De toute évidence, il s'agit d'une erreur d'assemblage lors du séquençage et donc d'une erreur de manip et non d'un nouveau variant. Le 11 janvier 2022, les séquences téléchargées furent retirées de GISAID, signifiant clairement qu'après vérification il s'agit vraisemblablement d'une contamination. Toutefois, selon une étude publiée sur "medXriv" le 28 mars 2022, deux cas d'infection par deux variants différents de Deltacron furent identifiés aux Etats-Unis. Selon les chercheurs, les coinfections par les variants Delta et Omicron et les évènements de recombinaison étaient encore rares. Les chercheurs ont séquencé 29719 échantillons positifs prélevés de novembre 2021 à février 2022, lorsque Delta et Omicron cocirculaient aux États-Unis. Ils ont trouvé 20 coinfections et deux cas indépendants d'infection par le Deltacron. Ils ont conclu que ce recombinant était rare et qu'il n'y avait actuellement aucune preuve que Deltacron était plus transmissible que les lignées Omicron déjà en circulation (BA.1, BA.2). Le sous-variant EG.5 d'Omicron Selon l'OMS, le sous-variant EG.5 d'Omicron fut signalé pour la première fois le 17 février 2023 et désigné comme variant sous surveillance (VUM) le 19 juillet 2023. Sa prévalence augmenta au niveau mondial, passant de 7.6% des cas de Covid-19 à la fin du mois de juin à 17.4% à la fin du mois de juillet. Par conséquent, il fut ensuite classé comme variant d'intérêt (VOI). Au 7 août 2023, il avait été identifié dans 51 pays et 7354 séquences génétiques avaient été soumises au GISAID. EG.5 est un sous-variant de XBB.1.9.2 d'Omicron qui présente le même profil d'acides aminés de la protéine S que le XBB.1.5 surnommé Kraken. Le sous-variant EG.5 porte une mutation supplémentaire F456L dans la protéine S par rapport au parent XBB.1.9.2. Au sein de la lignée EG.5, le sous-variant EG.5.1 surnommé "Eris" possède une mutation de pointe supplémentaire Q52H. Selon le professeur Andrew Pekosz du département de Microbiologie moléculaire et d'immunologie de l'Université Johns Hopkins, "les symptômes d'EG.5 semblent similaires à ceux d'autres variants" (fièvre, toux et fatigue, ainsi qu'un écoulement nasal, des maux de tête ou des douleurs musculaires. Ils peuvent ressembler à ceux d'un rhume, d'une grippe ou d'une pneumonie). En résumé, selon l'OMS, la gravité du sous-variant EG.5 n'est pas différente de celle des autres sous-variants d'Omicron circulant depuis fin 2021. Selon Andrew Pollard, professeur d'infectiologie et d'immunologie à l'Université d'Oxford, "il existe des preuves qu'Omicron et ses sous-variants sont moins graves que les souches antérieures du virus." Il ajoute "qu'étant donné que la population est très immunisée contre le SARS-CoV-2, notre immunité nous défendra contre les formes graves de la maladie." Les variants recombinants XD et XE (F, UK) Le 3 janvier 2022, un variant recombinant AY.4 de Delta et BA.1 d'Omicron fut identifié dans le nord de la France. L'OMS le nomma XD et est repris dans la base GISAID sous le nom de GKA. Un second variant recombinant BA.1 et BA.2 d'Omicron fut également identifié et nommé XE. Il est présent au Royaume-Uni où 1200 cas ont été enregistrés. Il serait 20% plus contagieux que XD. Le variant XD est un hybride de Delta porta des protéines S presque en totalité d'Omicron. Au 15 avril 2022, un total de 85 contaminations par le variant XD avaient été repertoriées dans cinq pays : la France, l'Allemagne, la Belgique, les Pays Bas et le Danemark (cf. BJM). Dans un article publié en pré-impression sur "Research Square" le 4 avril 2022, des chercheurs de l'institut Pasteur (F) ont présenté les résultats de la neutralisation du variant XD notamment. Ils ont fabriqué des pseudovirus possédant la protéine S du variant XD, Omicron ou Delta qui furent mis en contact avec du plasma sanguin de personnes vaccinées (2 doses, 1 dose + infection, 3 doses). Les anticorps dans les échantillons sanguins de personnes ayant reçu deux doses ont pu neutraliser le pseudovirus Delta mais il y avait très peu de neutralisation pour Omicron et XD. Dans les échantillons de personnes totalement vaccinées (3 doses), il y avait une neutralisation mais elle était 8 à 10 fois plus faible pour XD et Omicron que pour Delta. Cela montre que la protéine S d'Omicron offre au variant XD des capacités d’échappement face aux défenses immunitaires (naturelle ou après vaccination). Analyse des variants et de leurs effets Dans une étude publiée dans la revue "Cell" (en PDF) le 3 septembre 2020, Huang Weijin du NIFDC chinois et ses collègues ont constaté chez près d'une centaine de variants du SARS-CoV-2 la forte glycosylation de la protéine S; dans de nombreux sites de son génome, une mutation (N→Q) permet à un glucide de se lier à la protéine pour faciliter la réaction enzymatique. Les chercheurs ont étudié 80 variants et 26 mutations du SARS-CoV-2 et analysé leurs effets sur l'infectivité et la réactivité des anticorps neutralisants et les sérums d'anciens patients en convalescence.
Les schémas ci-dessus préparés par Weijin et ses collègues présentent les substitutions des acides aminés dans les variants qu'ils ont étudiés. Au-dessus, en rouge figurent les variants (D614G, D839Y, G1124V, L5F, L8V) et en vert les variants combinés avec D614G sur l'ensemble du gène codant pour la protéine S à l'exclusion du RBD (le domaine de liaison au récepteur). Au centre sont représentés les variants dans RBD. Enfin, en dessous, en bleu figurent toutes les mutations sur les sites supposés de glycosylation (N→Q), soit 22 sites. Ce groupe comprend des mutations (N→Q), une combinaison de deux mutations de site de glycosylation dans RBD et trois variants naturels (N74K, N149H et T719A), avec l'ablation des sites de glycosylation. Les sites où on observe fréquemment des substitution d'acides aminés (fréquence > 0.1%) sont surlignés en rouge. Selon les chercheurs, "le variant D614G, ainsi que plusieurs variants contenant à la fois D614G et une autre substitution d'acide aminé, étaient significativement plus infectieux. La plupart des variants présentant une mutation au niveau du domaine de liaison au récepteur étaient moins infectieux, mais des variants comprenant A475V, L452R, V483A et F490L sont devenus résistants à certains anticorps neutralisants. De plus, la majorité des délétions de glycosylation étaient moins infectieuses, tandis que la suppression de la glycosylation N331 et N343 réduisait considérablement l'infectivité, révélant l'importance de la glycosylation pour l'infectivité virale. Fait intéressant, le variant N234Q était très résistant aux anticorps neutralisants, tandis que le N165Q devenait plus sensible. Ces résultats pourraient être utiles pour le développement de vaccin et anticorps thérapeutiques." Concernant le variant Alpha (B.1.1.7), selon un rapport du comité SAGE publié le 21 janvier 2021, il serait associé à un risque plus élevé de décès que les autres variants. Au moins 6 variants du SARS-CoV-2 dont Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), Omicron (B.1.1.529) et B.1.1.28 sont à la fois plus contagieux et sont capables d'échapper aux défenses du système immunitaire. Ils sont également actifs plus longtemps. Mais rien ne prouve qu'il existerait un variant plus virulent que l'autre. En revanche, ils sont plus virulents que le SARS-CoV-2 original de Wuhan. Les virus recombinants du SARS-CoV-2 Dans un article publié sur le forum "Virological" le 18 décembre 2020, des chercheurs britanniques comprenant des membres du consortium COG-UK ont analysé des séquences génomiques du SARS-CoV-2 prélevées au Royaume-Uni et ont détecté un certain nombre de variants qui avaient été attribués au variant Alpha (lignée B.1.1.7) mais qui ne contiennent pas l'ensemble complet des mutations définissant le génome d'Alpha.
L'examen de ces séquences révéla que certaines sections du génome portent des mutations caractéristiques d'Alpha, tandis que d'autres sections portent des mutations spécifiques du variant B.1.429 originaire de Californie. Selon les chercheurs, "de longues séries de mutations le long du génome du SARS-CoV-2 qui correspondent à différentes lignées sont fortement indicatives de la recombinaison du virus". Lors d'une réunion (webinar) organisée par l'Académie des Sciences de New York le 2 février 2021, on apprit que le premier spécimen de virus recombinant du SARS-CoV-2 avait été découvert par la biologiste théoricienne et biophysicienne Bette Korber du Laboratoire National de Los Alamos (LANL) au Nouveau-Mexique qui déclara qu'elle en avait vu des preuves "assez claires" dans sa base de données de génomes viraux américains. Korber n'a vu qu'un seul génome recombinant parmi des milliers de séquences et à l'époque il n'était pas encore établi si le virus se transmettait d'une personne à l'autre ou s'il s'agissait de la mutation d'un virus ponctuel. Notons que sous la signature du rédacteur Graham Lawton, la revue "The New Scientist" du 16 février 2021 publia le premier article de vulgarisation sur cette découverte. Souligons que pour créer un vrai variant recombinant, l'un des variants dont il est composé doit circuler depuis longtemps et s'être propagé au sein d'une population suffisamment vaste. S'il ne sort de nul part, comme dans le cas du présumé Deltacron précité, il peut s'agi d'une erreur survenue en laboratoire. Puis, le 17 mars 2021 les mêmes chercheurs britanniques annoncèrent dans un nouvel article publié sur le forum "Virological" avoir identifié 8 virus recombinants du SARS-CoV-2. Ces versions hybrides combinent une partie du génome du variant Alpha et l'un des variants B.1.177, B.1.36.27, B.1.36.28 ou B.1.221, tous originaires majoritairement du Royaume-Uni (le même variant pouvant avoir été identifié dans plusieurs pays à la même époque). Ils sont répartis en quatre groupes auxquels s'ajoute 4 autres recombinants putatifs dont la liste est présentée ci-dessous. Ces virus recombinants se sont probablement développés au Royaume-Uni pendant une période de forte positivité au SARS-CoV-2, qui représentait ~1 à 2% de la population générale en Angleterre entre octobre 2020 et janvier 2021 (cf. UK - Office for National Statistics, 12 mars 2021). Pendant ce temps, le variant Alpha s'est rapidement répandu à travers le Royaume-Uni, remplaçant les variants britanniques précédemment existants qui présentaient déjà des incidences élevées, dont les plus courants étaient le B.1.177 et ses descendants (cf. E.Volz et al., 4 janvier 2021). Bien que moins fréquente que les variants, la recombinaison est une caractéristique fréquente et bien documentée de l'évolution moléculaire des coronavirus (cf. R.L. Graham et al., 2010) qu'on retrouve chez plusieurs espèces de Betacoronavirus du sous-genre Sarbecovirus (cf. M.F. Boni et al., et en PDF, 2020; B.Hu et al., 2017; C-C.Hon et al., 2008) dont le SARS-CoV-2 (cf. D.VanInsberghe et al., 2020). La recombinaison se produit probablement chez chaque individu contaminé, mais dans presque tous les cas, elle se produit entre des génomes presque identiques (descendants du petit pool de virus à l'origine de la contamination).
Pour rappel, ces recombinaisons sont le résultat d'une erreur lors de la copie de l'ARN viral par la polymérase. L'enzyme bloque sur un brin d'ARN et utilise à la place un autre brin qui s'ajoute à la nouvelle séquence. Selon les chercheurs, "ces remplacements d'acides aminés individuels dans le génome du SARS-CoV-2 sont principalement le résultat d'une réplication sujette aux erreurs et ne devraient pas avoir d'effets phénotypiques, la plupart des évènements de recombinaison sont simplement le résultat des processus intra-hôte qui se produisent à chaque contamination". Rappelons aussi que dans les vaccins recombinants, les chercheurs tirent parti de cette technique pour introduire dans les cellules humaines des vecteurs recombinés génétiquement qui permettront aux gènes de fabriquer des versions inoffensives de molécules étrangères dont la protéine S (cf. les vaccins Spoutnik V de Gamaleya, NVX-CoV2373 de Novavax, AZD1222 d'AstraZeneca/Oxford) afin que les défenses immunitaires apprennent à la reconnaître et la détruisent. S'il est possible de fabriquer un virus recombinant en laboratoire (cf. T.Fukuhara et al., 2020), jusqu'à preuve du contraire aucun n'est sorti des labos et n'a contaminé un être humain. Cette éventualité est donc exclue.
En revanche, on ne peut pas (encore) exclure une contamination survenue en laboratoire bien que la probabilité soit très faible. En effet, l'une des raisons possibles pour lesquelles une séquence génomique pourrait avoir une structure recombinée dite en mosaïque est qu'il s'agit du résultat d'un séquençage de lectures à partir de différents variants présents soit en raison d'une contamination en laboratoire, soit parce qu'un mélange de variants était présent dans l'échantillon ou dans le patient. Selon les chercheurs, bien que cela puisse donner naissance à des virus recombinants, le protocole de séquençage utilisé au Royaume-Uni (cf. J.R. Tyson et al., 2020) génère 98 amplicons courts de ~350 pb, de sorte que de longs domaines qui correspondent à un seul variant ou une seule lignée seraient peu probables. Pour les groupes A-D ci-dessus, cette possibilité peut également être exclue car plusieurs génomes ayant la même structure recombinée sont séquencés indépendamment de différents échantillons, ce qui implique qu'une transmission du recombinant s'est produite. Il convient également de noter que pour le groupe A, l'un des trois génomes recombinants putatifs fut séquencé dans une installation différente des deux autres. Pour les quatre autres virus recombinants putatifs ayant un seul représentant, selon les chercheurs il est possible que le cas échantillonné soit une coinfection et donc l'échantillon soit un mélange non naturel, mais vu le protocole de séquençage utilisé, c'est peu probable. Étant donné que les chercheurs ont observé quatre versions ou instances de génomes recombinants, ils concluent n'avoir aucune raison de soupçonner que les quatre singletons ne sont pas également des recombinants. Étant donné que depuis février ou mars 2021 de nombreux autres pays connaissent des niveaux d'incidence comparables à ceux du Royaume-Uni entre la fin de 2020 et le début de 2021, les chercheurs prévoient "que la recombinaison entre des lignées distinctes sera couramment détectée partout où un séquençage intensif du génome est entrepris. La recombinaison entre des variantes génétiquement similaires sera également courante mais plus difficile à détecter". Ces variants hybrides ne signifient pas nécessairement que le virus est plus contagieux ou plus virulent. Contrairement à une mutation, comme celle du variant Alpha (B.1.1.7) où les changements s'accumulent un par un, la recombinaison peut rassembler plusieurs mutations en une seule fois. La plupart du temps, si celles-ci ne confèrent aucun avantage au virus, parfois elles lui permettent de mieux s'adapter à son environnement. Selon François Balloux de l'University College London, la recombinaison peut avoir une importance évolutive majeure et est un phénomène considéré par beaucoup de spécialistes comme à l'origine du SARS-CoV-2. En effet, les zoonoses nous prouvent tous les jours que de nombreuses chauves-souris portent des virus recombinants de coronavirus. En théorie, la recombinaison pourrait conduire à l'émergence de nouveaux variants plus dangereux, mais actuellement on ne sait pas encore à quel point ces évènements de recombinaisons pourraient représenter une menace. Des prédispositions héritées des Néandertaliens La paléogénétique nous apprend que nous avons hérité d'une partie du patrimoine génétique des premiers humains (Homo sapiens et Homo denisova parmi d'autres) et de toute évidence d'autres sous-espèces par métissage. En effet, certaines populations ont hérité de nos ancêtres de gènes spécifiques leur permettant par exemple de mieux supporter les climats extrêmes ou renforçant leur système immunitaire face à certaines maladies. Mais parfois, cet héritage cause plus de mal que de bien. Des études ont montré que certaines mutations génétiques sur le chromosome 3 étaient fréquemment présentes chez les patients Covid souffrant de formes sévères de Covid-19. A ce jour, on a identifié 22 variants génétiques ou allèles touchant 8 gènes différents ayant un effet délétère sur la réponse du système immunitaire contre le Covid-19. Près de 15% des formes sévères de Covid s'expliquent par ces prédispositions génétiques héritées des Néandertaliens. En revanche, d'autres variants génétiques (locus OSA) présents sur le chromosome 12 des population non africaines réduisent de ~22% le risque de développer une forme sévère de la Covid-19. Nous reviendrons en détails sur le sujet dans l'article consacré au métissage entre les premiers humains et leurs conséquences actuelles. Dernier chapitre |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||